

Key Points
HRG attenuates polyP-induced thrombin generation.
Thrombosis induced in mice by intraperitoneal injection of polyP is mediated by factor XII and attenuated by HRG.

Abstract
Histidine-rich glycoprotein (HRG) is an abundant plasma protein that binds factor XIIa (FXIIa) and inhibits factor XII (FXII) autoactivation and FXIIa-mediated activation of FXI. Polyphosphate (polyP), a potent procoagulant released from activated platelets, may serve as a physiological activator of the contact system. Previously, we showed that HRG binds DNA and neutralizes its procoagulant activity. Consequently, our goal was to determine whether the capacity of HRG to bind polyanions enables it to regulate polyP-induced thrombosis. In a plate-based assay, immobilized polyP bound HRG, FXII, and FXIIa in a zinc-dependent manner. Basal and polyP-induced thrombin generation was greater in plasma from HRG-deficient mice than in plasma from wild-type mice. Intraperitoneal injection of polyP shortened the activated partial thromboplastin time, enhanced thrombin generation, increased thrombin-antithrombin levels, reduced lung perfusion, and promoted pulmonary fibrin deposition to a greater extent in HRG-deficient mice than in wild-type mice, effects that were abrogated with FXII knockdown. HRG thus attenuates the procoagulant and prothrombotic effects of polyP in an FXII-dependent manner by modulating the contact system.
Introduction
The contact system is initiated when factor XII (FXII) binds to negatively charged surfaces and undergoes autoactivation.1-3 Such surfaces include the naturally occurring polyanions DNA, RNA, and inorganic polyphosphates (polyPs).2,4 DNA and RNA are released from activated or damaged cells, whereas polyP is released from activated platelets.2,4,5 Activation of FXII by polyanions propagates a procoagulant response that promotes thrombin generation and fibrin formation and leads to thrombosis.2,4
polyP is a linear polymer of inorganic phosphate units held together by phosphoanhydride bonds.5,6 polyP is abundant in nature and is present in prokaryotes, lower eukaryotes, and vertebrates.7 In mammals, polyP secreted from the dense granules of activated platelets is composed of 60 to 100 phosphate units.8,9 In addition to promoting FXII autoactivation, platelet polyP enhances the activation of factor XI and factor V by thrombin and increases fibrin clot stability, thereby attenuating fibrin degradation.8,10,11 Therefore, polyP is an important contributor to the procoagulant effect of platelets.
Histidine-rich glycoprotein (HRG) is a 75 kDa glycoprotein that is synthesized in the liver and circulates in plasma at a concentration of 1 to 3 μM.12 HRG has been implicated in a variety of biological processes, including angiogenesis, immunity, and coagulation.12-15 HRG levels are reduced in pregnancy and with sepsis, indicating that it is a negative acute-phase reactant protein.16 Previously, we showed that HRG downregulates the contact system by binding factor XIIa (FXIIa) with high affinity and attenuating FXII autoactivation and FXIIa-mediated activation of FXI.17 HRG binds FXIIa at a site distinct from the active site because FXIIa chromogenic activity and its inhibition by corn trypsin inhibitor (CTI) are unaffected. The importance of this regulation is highlighted by the finding that FeCl3-induced arterial thrombosis is accelerated in mice deficient in HRG compared with that in wild-type mice.18 These observations suggest that HRG serves as a molecular brake for the contact system.
Because HRG binds DNA and RNA and attenuates their procoagulant activity,18 we hypothesized that HRG could also bind polyP and attenuate its procoagulant activity. To test this hypothesis, we measured the affinity of HRG for polyP, examined the effect of HRG on the procoagulant activity of polyP in vitro, and compared the prothrombotic effects of polyP in HRG-deficient and wild-type mice with or without FXII knockdown.
Methods
Materials
A detailed description of the study materials is provided in the supplemental Methods. Short-chain polyP of ∼70 phosphate units generously provided by Thomas Staffel (70.5 CAREPHOS 244, BK Giulini, GmbH, Ladenburg, Germany), was dissolved in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.4 for the in vitro studies and in 0.9% sterile saline for in vivo without added calcium.
Affinity of polyP for HRG, FXII, and FXIIa
An enzyme-linked immunosorbent assay was used to determine the affinity of HRG, FXII, and FXIIa for immobilized biotinylated (b)-polyP.19 polyP of ∼70 phosphate units was biotinylated as described in the supplemental Methods. In the wells of a 96-well Costar high-binding enzyme-linked immunosorbent assay plate (Corning, Tewksbury, MA), 10 μg/mL streptavidin in 50 mM sodium carbonate buffer, pH 9.6 was incubated overnight at 4°C. Wells were then blocked with phosphate-buffered saline, pH 7.4 containing 3% bovine serum albumin (BSA) for 90 minutes at 23°C and then washed 3 times with wash buffer consisting of 20 mM Tris-HCl, 150 mM NaCl, pH 7.4 containing 0.01% Tween 20. After the addition of 10 μg/mL (100 μM) b-polyP to each well, the plate was incubated overnight at 4°C, and wells were washed 3 times. To the wells, varying concentrations of HRG, FXII, or FXIIa were added in 20 mM HEPES, 150 mM NaCl, pH 7.4 containing 1% BSA and 0.05% Tween 20 in the absence or presence of 6 μM ZnCl2. After incubation for 2 hours at 23°C, wells were washed 3 times. Bound HRG and BSA were detected with a 1:200 dilution of peroxidase-labeled sheep anti-human HRG antibody, whereas bound FXII and FXIIa were detected with a 1:100 dilution of peroxidase-labeled goat anti-human FXII antibody. After incubation for 2 hours at 23°C and 3 washes, 0.4 mg/mL o-phenylenediamine dihydrochloride in substrate buffer (50 mM citric acid and 50 mM disodium phosphate, pH 5.0), containing 30% H2O2 was added to each well. Reactions were stopped after 10 to 15 minutes with 0.83 M H2SO4, and absorbance was measured at 490 nm by using a SpectraMax M3 plate reader (Molecular Devices, San Jose, CA).
Data were analyzed by using nonlinear regression of a rectangular hyperbola using TableCurve 2D (Jandel Scientific, Systat Software Inc., San Jose, CA) to determine the dissociation constants (Kd).
Mice
HRG−/− mice were maintained on a pure C57BL/6 (The Jackson Laboratory, Bar Harbor, ME) background after >10 generations of backcrossing. Mice were housed in micro-isolator cages exposed to 12-hour light-dark cycles and had free access to food and water. Blood was collected and plasma was prepared as described in the supplemental Methods. Age- and weight-matched male mice were used for all experiments. All animal utilization protocols were approved by the Animal Research Ethics Board at McMaster University, and studies were performed in accordance with the Canadian Council on Animal Care guidelines.
FXII was knocked down in wild-type and HRG−/− mice with a liver-directed, mouse-specific FXII–antisense oligonucleotide (ASO), which was generously provided by Ionis Pharmaceuticals (Carlsbad, CA), as previously described, with some modifications.20 Briefly, the FXII-ASO was administered subcutaneously at a dose of 20 mg/kg twice weekly for 4 weeks, and the extent of knockdown was assessed by comparing hepatic FXII messenger RNA and plasma FXII levels before and after ASO administration, as described in the supplemental Methods. Mice were studied after 4 weeks of ASO treatment.
polyP-induced thrombosis model
HRG−/− and wild-type mice were anesthetized by using a combination of oxygen (0.5 L/min) and isoflurane (1-4%) delivered by face mask, and the skin of the abdomen was aseptically prepared. polyP (100 mg/kg in saline) or an equivalent volume of saline was given with an intraperitoneal (IP) injection using an insulin syringe fitted with a 30 G needle. Mice were euthanized via cervical dislocation at varying time points. Blood was collected via puncture of the inferior vena cava and plasma was prepared as described in the supplemental Methods. Lungs, liver, heart, and kidneys were removed for histologic analysis as detailed in the supplemental Methods.
Coagulation testing and thrombin generation assays
To evaluate the impact of polyP and HRG on clotting in human and murine plasma, clotting assays were conducted as described in the supplemental Methods.
To monitor thrombin generation, 40 μL of mouse plasma was incubated with 15 μM of phosphatidylcholine-phosphatidylserine (3:1 mol:mol) vesicles (PCPS) for 10 minutes at 37°C before initiating thrombin generation by addition of 50 μL of a solution containing 15 mM CaCl2 and 1 mM Z-Gly-Gly-Arg-AMC. Substrate hydrolysis was monitored by using a SpectraMax M3 plate reader (Molecular Devices) for 90 minutes at excitation and emission wavelengths of 360 nm and 460 nm, respectively. Thrombin generation profiles were analyzed by using Technothrombin TGA software (Technoclone, Vienna, Austria). To determine the functional concentration of polyP absorbed after IP injection, peak thrombin levels in plasma measured at various time points were compared with a standard curve produced by adding known concentrations of polyP to pooled plasma from wild-type or HRG−/− mice.
To confirm that polyP was responsible for the procoagulant effects, plasma from wild-type or HRG−/− mice treated with IP polyP was incubated with 0 to 20 U/mL of calf intestinal alkaline phosphatase (Promega, Madison, WI) for 10 minutes at 37°C before measuring clotting times and thrombin generation.
Evans blue dye lung perfusion
In one-half of the mice, Evans blue dye was used to assess lung perfusion 1 hour after IP injection of polyP or saline. Briefly, 200 μL of 1% sterile-filtered Evans blue dye solution was injected into the right ventricle over 30 seconds using a syringe pump (Harvard Apparatus, Holliston, MA).21-23 The right atrium was then opened, and the remaining dye was flushed from the right ventricle by injection of 2 mL saline over 1 minute by using the syringe pump. The lungs and heart were collected, rinsed with water for 1 minute, and then imaged at standardized exposure settings with a white background. After weighing each lung, Evans blue dye was extracted by incubating the lungs in formamide overnight at 55°C.21,22 The absorbance of the supernatants was then determined at 610 nm and compared with that of known amounts of dye to calculate the concentration of dye extracted per milligram of lung tissue.21
Quantification of thrombin-antithrombin complexes
Thrombin-antithrombin levels were quantified by immunoassay (Affinity Biologicals, Hamilton, ON, Canada) with modifications as described in the supplemental Methods.
Statistical analysis
All data are presented as mean ± standard deviation. Significance of differences between groups was determined by using one- or two-way analysis of variance followed by the Holm- Šídák comparison test. SigmaPlot (Systat Software, San Jose, CA) and IBM SPSS Statistics software (Armonk, NY) were used to create graphs and perform the statistical analyses, respectively. For all analyses, P values <.05 were considered significant.
Results
HRG, FXII, and FXIIa bind polyP
Previous studies have shown that HRG binds various polyanions with high affinity, including DNA and heparin.12,24 To examine whether HRG also binds polyP, b-polyP was immobilized in the wells of a streptavidin-coated multi-well plate, and binding of HRG, FXII, or FXIIa was assessed. BSA was used as a control. Whereas no BSA binding was detected, HRG bound b-polyP with a Kd value of 152 ± 9 nM, while FXII and FXIIa bound b-polyP with Kd values of 104 ± 25 nM and 97 ± 13 nM, respectively (Figure 1A). All interactions were observed in the presence of 6 µM ZnCl2 but not in its absence. These results indicate that, similar to DNA and RNA, polyP binds HRG, FXII, and FXIIa in a zinc-dependent manner.18

Interaction of HRG, FXII, and FXIIa with polyP and the effects of HRG on polyP-induced clotting. (A) b-polyP was immobilized in wells of a streptavidin-coated multi-well plate, and binding of BSA, HRG, FXII, and FXIIa was quantified in the absence or presence of 6 μM ZnCl2. Absorbance values for the associated ligand concentrations are plotted; the data were analyzed by nonlinear regression of a rectangular hyperbola (lines). Symbols represent mean ± standard deviation (SD) of 5 determinations. (B) Clotting after recalcification of control (closed circles with solid red line) or HRG-depleted human plasma (open circles with dashed blue line) in the absence or presence of polyP at the indicated concentrations was monitored by absorbance, and the clot time was calculated as the time to achieve half-maximum absorbance. Lines represent nonlinear regression analysis of the data. Symbols represent mean ± SD of 5 determinations. (C) polyP-induced clotting times in control (red bar) and HRG-depleted human plasma (blue bars) containing human HRG at the indicated concentrations were determined. Bars represent mean ± SD of 4 determinations. *P < .05, **P < .01 compared with normal pooled control plasma. (D) Control and HRG-depleted plasma were incubated with or without 1 μM CTI for 15 minutes followed by the addition of either aPTT-SP reagent, RecombiPlasTin reagent (Instrumentation Laboratory, Bedford, MA), or 40 μg/mL of polyP. Clotting was initiated with 26 mM CaCl2, and the clot time was calculated as the time to half maximum increase in absorbance. Bars represent mean ± SD of 4 determinations each done in duplicate. *P < .05, ****P < .001 comparison between control and HRG-depleted plasma as indicated by the lines. NS, not significant (analysis of variance, Holm-Šídákmethod); PT, prothrombin time.
Interaction of HRG, FXII, and FXIIa with polyP and the effects of HRG on polyP-induced clotting. (A) b-polyP was immobilized in wells of a streptavidin-coated multi-well plate, and binding of BSA, HRG, FXII, and FXIIa was quantified in the absence or presence of 6 μM ZnCl2. Absorbance values for the associated ligand concentrations are plotted; the data were analyzed by nonlinear regression of a rectangular hyperbola (lines). Symbols represent mean ± standard deviation (SD) of 5 determinations. (B) Clotting after recalcification of control (closed circles with solid red line) or HRG-depleted human plasma (open circles with dashed blue line) in the absence or presence of polyP at the indicated concentrations was monitored by absorbance, and the clot time was calculated as the time to achieve half-maximum absorbance. Lines represent nonlinear regression analysis of the data. Symbols represent mean ± SD of 5 determinations. (C) polyP-induced clotting times in control (red bar) and HRG-depleted human plasma (blue bars) containing human HRG at the indicated concentrations were determined. Bars represent mean ± SD of 4 determinations. *P < .05, **P < .01 compared with normal pooled control plasma. (D) Control and HRG-depleted plasma were incubated with or without 1 μM CTI for 15 minutes followed by the addition of either aPTT-SP reagent, RecombiPlasTin reagent (Instrumentation Laboratory, Bedford, MA), or 40 μg/mL of polyP. Clotting was initiated with 26 mM CaCl2, and the clot time was calculated as the time to half maximum increase in absorbance. Bars represent mean ± SD of 4 determinations each done in duplicate. *P < .05, ****P < .001 comparison between control and HRG-depleted plasma as indicated by the lines. NS, not significant (analysis of variance, Holm-Šídákmethod); PT, prothrombin time.
HRG attenuates polyP-induced clotting in vitro
To determine the impact of HRG on polyP-initiated clotting in vitro, we compared its effect on clotting times in control and HRG-depleted human plasma. Depletion of HRG from human plasma from immunoaffinity chromatography was confirmed by immunoblotting (supplemental Figure 1). polyP shortened the recalcification time in control and HRG-depleted plasma in a concentration-dependent manner (Figure 1B). With 40 µg/mL polyP, the recalcification time was significantly shorter in HRG-depleted plasma than in control plasma (129 ± 11 seconds and 224 ± 18 seconds, respectively; P < .05). Addition of human HRG to HRG-depleted plasma attenuated polyP-induced clotting by up to twofold, confirming that HRG modulates the procoagulant activity of polyP (Figure 1C). At physiological concentrations (1-2 µM), HRG attenuated clotting in HRG-depleted plasma to an extent comparable with that of control plasma.
When incubated with FXII, polyP promoted FXII autoactivation in a concentration-dependent manner in the presence of zinc but not in its absence (supplemental Figure 2A). Activation of FXII was confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis analysis (supplemental Figure 2B). Because HRG binds FXIIa with high affinity and inhibits polyP-induced FXII autoactivation with a 50% inhibitory concentration value of 232 ± 25 nM (supplemental Figure 2C), we examined the role of FXII in polyP-induced clotting by examining the effect of CTI, a potent and specific inhibitor of FXII, on polyP-induced clotting. CTI significantly reduced the procoagulant activity of polyP to background levels and prolonged the activated partial thromboplastin time (aPTT) in control and HRG-depleted plasma but had no effect on the prothrombin time (Figure 1D). These results confirm that polyP enhances clotting in an FXII-dependent manner in vitro and show that this activity is attenuated by HRG.
Procoagulant activity of polyP is greater in HRG-deficient mice than in wild-type mice
To determine whether HRG modulates the procoagulant activity of polyP in vivo, wild-type and HRG−/− mice were given an IP injection of polyP (100 mg/kg) or saline, and clotting times and thrombin generation were measured in plasma harvested from blood samples collected at various time points thereafter. Compared with saline, IP polyP progressively shortened the plasma recalcification time for up to 60 minutes after injection (Figure 2A). Thus, compared with saline, polyP shortened the clotting time by 5.2-fold in wild-type mice (from 803 ± 41 seconds to 153 ± 7 seconds; P < .001) and by 7.4-fold in HRG−/− mice (from 667 ± 33 seconds to 90 ± 16 seconds; P < .001) at 60 minutes, suggesting that the procoagulant effect of polyP is greater in HRG−/− mice than in wild-type mice.
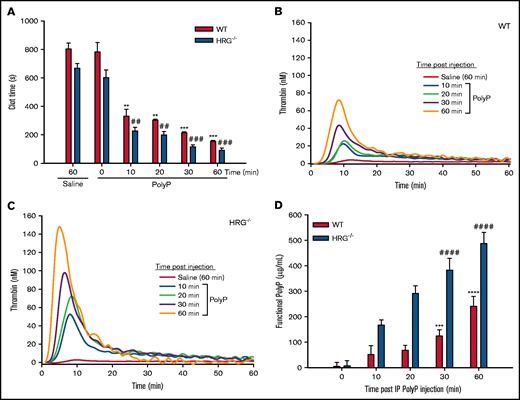
Effect of IP polyP on plasma clotting and thrombin generation in wild-type (WT) and HRG-deficient mice. (A) Clotting times were determined by recalcification of plasma obtained from WT or HRG−/− mice administered IP saline or polyP. (B and C) Thrombin generation was determined in plasma from saline- or polyP-treated WT or HRG−/− mice collected at the indicated time points. Thrombin generation was initiated with 15 mM CaCl2 in the presence of 15 µM of phosphatidylcholine-phosphatidylserine vesicles and was quantified by monitoring hydrolysis of 1 mM Z-Gly-Gly-Arg-AMC thrombin substrate. (D) Functional polyP concentrations in plasma at various time points after IP injections of polyP were quantified in a thrombin generation assay by comparing peak thrombin concentrations at the sampling times shown with a standard curve of peak thrombin concentrations generated by adding known amounts of polyP to plasma from WT or HRG−/− mice. N = 9 mice per group; bars represent mean ± standard deviation. **P < .01, ***P < .001, ****P < .0001 compared with saline-treated WT mice; ##P < .01, ###P < .001, ####P < .0001 compared with saline treated HRG−/− mice (analysis of variance, Holm-Šídákmethod).
Effect of IP polyP on plasma clotting and thrombin generation in wild-type (WT) and HRG-deficient mice. (A) Clotting times were determined by recalcification of plasma obtained from WT or HRG−/− mice administered IP saline or polyP. (B and C) Thrombin generation was determined in plasma from saline- or polyP-treated WT or HRG−/− mice collected at the indicated time points. Thrombin generation was initiated with 15 mM CaCl2 in the presence of 15 µM of phosphatidylcholine-phosphatidylserine vesicles and was quantified by monitoring hydrolysis of 1 mM Z-Gly-Gly-Arg-AMC thrombin substrate. (D) Functional polyP concentrations in plasma at various time points after IP injections of polyP were quantified in a thrombin generation assay by comparing peak thrombin concentrations at the sampling times shown with a standard curve of peak thrombin concentrations generated by adding known amounts of polyP to plasma from WT or HRG−/− mice. N = 9 mice per group; bars represent mean ± standard deviation. **P < .01, ***P < .001, ****P < .0001 compared with saline-treated WT mice; ##P < .01, ###P < .001, ####P < .0001 compared with saline treated HRG−/− mice (analysis of variance, Holm-Šídákmethod).
At 60 minutes after IP saline or polyP administration, the aPTT in HRG−/− mice was significantly shorter than that in wild-type mice (259 ± 29 seconds and 218 ± 15 seconds, respectively; P < .05) (supplemental Figure 3A). Thus, polyP significantly shortened the aPTT by threefold in HRG−/− mice and by twofold in wild-type mice. In contrast, the prothrombin time values in HRG−/− and wild-type mice administered polyP were not significantly different from those in mice administered saline (supplemental Figure 3).
Whereas there was minimal thrombin generation in recalcified plasma from HRG−/− or wild-type mice 60 minutes after saline administration, there was a progressive increase in thrombin generation after IP polyP administration (Figure 2B-C). The peak thrombin concentration was twofold higher in HRG−/− mice than in wild-type mice after 60 minutes. Likewise, the lag time and time to peak thrombin were significantly shorter, and endogenous thrombin potential was greater in plasma from HRG−/− mice than in plasma from wild-type mice (supplemental Figure 3C-F).
The concentration of IP polyP absorbed into the bloodstream was quantified in a functional assay by comparing peak thrombin concentrations with a standard curve created by adding known concentrations of polyP to plasma from untreated HRG−/− or wild-type mice. One hour after injection, the functional concentration of procoagulant polyP detected in plasma significantly increased to a maximum of 487 ± 43 µg/mL in HRG−/− mice and 241 ± 38 µg/mL in wild-type mice (Figure 2D). Two hours after polyP administration, the functional polyP concentrations significantly decreased by 1.2 fold in HRG−/− mice and 1.9-fold in wild-type mice (supplemental Figure 4), consistent with the reported plasma half-life of polyP of 1.5 to 2 hours.6,25 Thus, the procoagulant effect of polyP is greater in plasma from HRG−/− mice than from wild-type mice, consistent with the lack of HRG.
Alkaline phosphatase was used to confirm that polyP was responsible for the observed procoagulant effects. Thus, thrombin generation and clotting times were measured before and after the addition of alkaline phosphatase to plasma obtained from wild-type or HRG−/− mice 60 minutes after polyP administration. Alkaline phosphatase attenuated thrombin generation (Figure 3A-B) and prolonged plasma clotting times in a concentration-dependent manner (Figure 3C). Degradation of polyP by alkaline phosphatase was confirmed by polyacrylamide gel electrophoresis analysis (Figure 3D). These findings suggest that polyP injected intraperitoneally is absorbed into the circulation unchanged and that the absorbed polyP is responsible for the shortening of the recalcification time and the aPTT and the enhancement of thrombin generation.
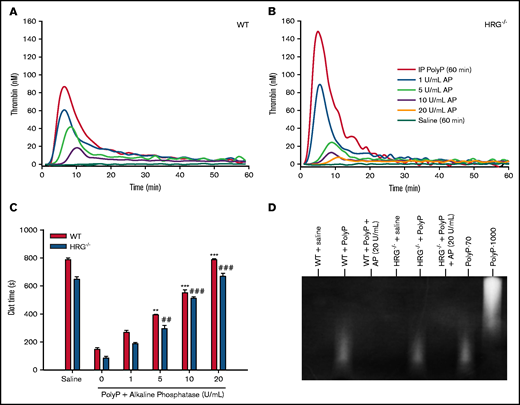
Effect of alkaline phosphatase (AP) on clotting times and thrombin generation in plasma from wild-type (WT) or HRG-deficient mice. (A and B) AP was incubated for 15 minutes at 37°C with plasma from WT or HRG−/− mice collected 60 minutes after IP polyP injection. Thrombin generation was initiated by adding 15 mM CaCl2 and was quantified by monitoring the hydrolysis of 1 mM Z-GGR-AMC thrombin substrate. N = 6 mice per group. (C) Plasma recalcification times were determined in plasma collected from WT or HRG−/− mice 60 minutes after IP polyP administration with or without preincubation with AP. Absorbance was measured at 405 nm for 1 hour, and the clot time was determined as the time to half maximal absorbance. N = 6 mice per group; bars represent mean ± standard deviation. (D) Plasma samples from mice injected with polyP before and after AP treatment were subjected to electrophoresis on a TBE-urea gel, and polyP was imaged by negative staining with 4′,6-diamidino-2-phenylindole. PolyP composed of either 70 or 1000 phosphate units (polyP-70 and polyP-1000, respectively) was used as standards. Representative images from 6 gels per treatment group are shown. **P < .01, ***P < .001 compared with plasma from WT mice without AP; ##P < .01, ###P < .001 compared with plasma from HRG−/− mice without AP (analysis of variance, Holm-Šídákmethod).
Effect of alkaline phosphatase (AP) on clotting times and thrombin generation in plasma from wild-type (WT) or HRG-deficient mice. (A and B) AP was incubated for 15 minutes at 37°C with plasma from WT or HRG−/− mice collected 60 minutes after IP polyP injection. Thrombin generation was initiated by adding 15 mM CaCl2 and was quantified by monitoring the hydrolysis of 1 mM Z-GGR-AMC thrombin substrate. N = 6 mice per group. (C) Plasma recalcification times were determined in plasma collected from WT or HRG−/− mice 60 minutes after IP polyP administration with or without preincubation with AP. Absorbance was measured at 405 nm for 1 hour, and the clot time was determined as the time to half maximal absorbance. N = 6 mice per group; bars represent mean ± standard deviation. (D) Plasma samples from mice injected with polyP before and after AP treatment were subjected to electrophoresis on a TBE-urea gel, and polyP was imaged by negative staining with 4′,6-diamidino-2-phenylindole. PolyP composed of either 70 or 1000 phosphate units (polyP-70 and polyP-1000, respectively) was used as standards. Representative images from 6 gels per treatment group are shown. **P < .01, ***P < .001 compared with plasma from WT mice without AP; ##P < .01, ###P < .001 compared with plasma from HRG−/− mice without AP (analysis of variance, Holm-Šídákmethod).
Prothrombotic activity of polyP is greater in HRG-deficient mice than in wild-type mice
To assess the prothrombotic effects of polyP, pulmonary perfusion 60 minutes after IP polyP injection was evaluated by using Evans blue dye, and lung sections were subjected to immunofluorescence analysis to detect fibrin accumulation.21,22 Visual inspection of the lungs suggested greater impairment of lung perfusion in HRG−/− mice than in wild-type mice (Figure 4A), a finding confirmed by quantifying the dye extracted from the lungs. Specifically, polyP administration completely impaired perfusion in HRG−/− mice because the lungs remained pink, consistent with vascular occlusion. These results were confirmed by quantifying the dye extracted from the lungs. Thus, in wild-type mice, the dye extracted decreased from 3.6 ± 0.07 µg/mg with saline to 1.7 ± 0.3 µg/mg with polyP (P < .001) (Figure 4B). In HRG−/− mice, the dye extracted decreased from 2.9 ± 0.1 µg/mg with saline to 0.6 ± 0.04 µg/mg with polyP (P < .01). Notably, with both saline and polyP administration, perfusion was lower in HRG−/− mice than in wild-type mice, raising the possibility that baseline pulmonary fibrin deposition is greater in HRG−/− mice than in wild-type mice. This concept was confirmed by quantification of fibrin deposition by immunofluorescence (Figure 4C-D). Fibrin deposition was scored in a blinded manner by 2 analysts yielding a strong κ agreement of 0.86 (P < .001). With saline treatment, HRG−/− mice exhibited significantly more fibrin deposition than wild-type mice (P < .05), suggesting that HRG deficiency is associated with a prothrombotic phenotype. With polyP injection, fibrin deposition in the lungs at 60 minutes was twofold higher (P < .001) in HRG−/− mice than in wild-type mice. Examination of sections from kidney, heart, and liver tissue showed no significant differences in fibrin deposition after polyP administration in HRG−/− vs wild-type mice (data not shown). To confirm the immunofluorescence results, fibrin deposition in whole lung lysates was quantified by western blotting. Compared with wild-type mice, HRG−/− mice exhibited significantly enhanced fibrin deposition in the lung lysate samples after polyP administration (supplemental Figure 5). Together, these results suggest that the prothrombotic activity of polyP is enhanced in HRG-deficient mice.
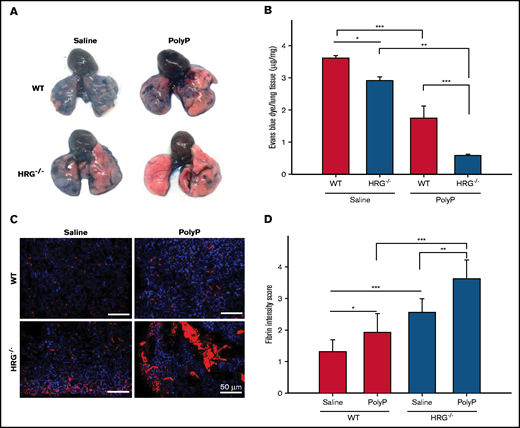
Comparison of the effect of IP polyP on perfusion and fibrin deposition in the lungs of wild-type (WT) or HRG-deficient mice. (A) Evans blue dye was injected into the right ventricle of WT or HRG−/− mice administered IP saline or polyP. Images of the lungs were taken to examine the extent of dye perfusion into the lungs. Representative images from 6 mice per group are shown. (B) Evans blue dye extracted from lungs with formamide was quantified by measuring absorbance at 605 nm and calculating the amount of dye per microgram of tissue by comparison with a standard curve. N = 6 mice per group; bars represent mean ± standard deviation. (C) Fibrin deposition (red) in lung sections was detected by immunofluorescence with 4′,6-diamidino-2-phenylindole (blue) serving as a nuclear stain. Scale bars represent 50 μm. Representative images from six mice per treatment group are shown. (D) Fibrin intensity was scored in a blinded manner by using a scale of 0 to 4, with 4 representing the greatest intensity. N = 6 mice per group; bars represent mean ± standard deviation. *P < .05, **P < .01, ***P < .001 comparison between saline- or polyP-treated WT or HRG−/− mice as indicated by the lines (analysis of variance, Holm-Šídákmethod).
Comparison of the effect of IP polyP on perfusion and fibrin deposition in the lungs of wild-type (WT) or HRG-deficient mice. (A) Evans blue dye was injected into the right ventricle of WT or HRG−/− mice administered IP saline or polyP. Images of the lungs were taken to examine the extent of dye perfusion into the lungs. Representative images from 6 mice per group are shown. (B) Evans blue dye extracted from lungs with formamide was quantified by measuring absorbance at 605 nm and calculating the amount of dye per microgram of tissue by comparison with a standard curve. N = 6 mice per group; bars represent mean ± standard deviation. (C) Fibrin deposition (red) in lung sections was detected by immunofluorescence with 4′,6-diamidino-2-phenylindole (blue) serving as a nuclear stain. Scale bars represent 50 μm. Representative images from six mice per treatment group are shown. (D) Fibrin intensity was scored in a blinded manner by using a scale of 0 to 4, with 4 representing the greatest intensity. N = 6 mice per group; bars represent mean ± standard deviation. *P < .05, **P < .01, ***P < .001 comparison between saline- or polyP-treated WT or HRG−/− mice as indicated by the lines (analysis of variance, Holm-Šídákmethod).
HRG modulates polyP-induced thrombosis in an FXII-dependent manner
Because polyP is known to activate the contact system,10,26 we examined the role of FXII in this process by knocking down the levels of FXII in HRG−/− and wild-type mice using a mouse-specific ASO.20 ASO treatment reduced FXII messenger RNA by >90% and lowered plasma FXII to undetectable levels according to western blot analysis (supplemental Figure 6A-B). Treatment with the FXII-ASO significantly prolonged the aPTT in wild-type and HRG−/− mice but had no effect on the prothrombin time (supplemental Figure 6C-D). Compared with its effects in mice with normal levels of FXII, polyP had little effect on thrombin generation or the plasma recalcification time when FXII was knocked down (Figure 5A-C). Levels of thrombin-antithrombin complexes after polyP administration were significantly lower with FXII knockdown than without (Figure 5D), and FXII knockdown reduced fibrin deposition in the lungs to levels similar to that with saline administration (Figure 5E-F). Together, these results suggest that polyP-induced thrombosis in HRG−/− and wild-type mice is FXII dependent.
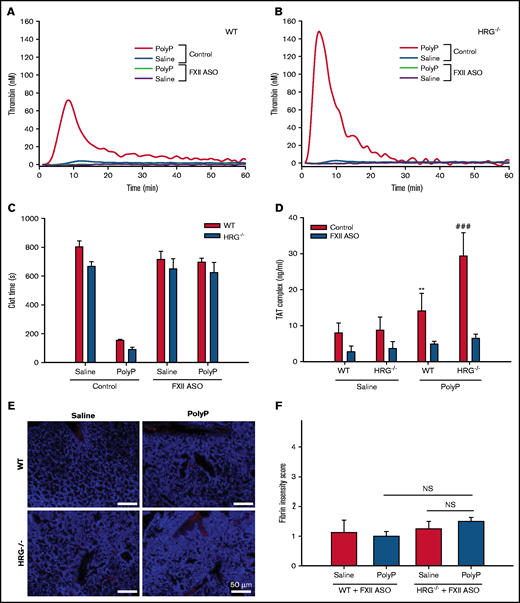
Effect of FXII knockdown on the procoagulant and prothrombotic effects of polyP in wild-type (WT) or HRG-deficient mice. Thrombin generation was measured in plasma collected 60 minutes after IP saline or polyP treatment of WT mice (A) or HRG−/− mice (B) without (control) or with FXII knockdown by ASO treatment. N = 8 mice per group. (C) Recalcification times were measured in plasma collected 60 minutes after WT or HRG−/− mice without or with FXII knockdown were given IP saline or polyP. N = 8 mice per group; bars represent mean ± standard deviation (SD). (D) Thrombin-antithrombin (TAT) complex levels in plasma from WT or HRG−/− mice without (red bars) or with FXII knockdown (blue bars). N = 8 to 9 mice per group; bars represent mean ± SD. (E) Fibrin deposition (red) in lungs from WT or HRG−/− mice without or with FXII knockdown was identified by immunofluorescence using an anti-fibrin(ogen) antibody. 4′,6-Diamidino-2-phenylindole (blue) was used as a nuclear stain. Scale bar represents 50 μm. (F) Fibrin intensities in panel E were quantified in a blinded manner by using a scale of 0 to 4. N = 8 mice per group; bars represent mean ± SD. **P < .01 compared with WT mice with FXII knockdown given saline; ###P < .001 compared with HRG−/− mice with FXII knockdown given saline (analysis of variance, Holm-Šídák method). NS, not significant.
Effect of FXII knockdown on the procoagulant and prothrombotic effects of polyP in wild-type (WT) or HRG-deficient mice. Thrombin generation was measured in plasma collected 60 minutes after IP saline or polyP treatment of WT mice (A) or HRG−/− mice (B) without (control) or with FXII knockdown by ASO treatment. N = 8 mice per group. (C) Recalcification times were measured in plasma collected 60 minutes after WT or HRG−/− mice without or with FXII knockdown were given IP saline or polyP. N = 8 mice per group; bars represent mean ± standard deviation (SD). (D) Thrombin-antithrombin (TAT) complex levels in plasma from WT or HRG−/− mice without (red bars) or with FXII knockdown (blue bars). N = 8 to 9 mice per group; bars represent mean ± SD. (E) Fibrin deposition (red) in lungs from WT or HRG−/− mice without or with FXII knockdown was identified by immunofluorescence using an anti-fibrin(ogen) antibody. 4′,6-Diamidino-2-phenylindole (blue) was used as a nuclear stain. Scale bar represents 50 μm. (F) Fibrin intensities in panel E were quantified in a blinded manner by using a scale of 0 to 4. N = 8 mice per group; bars represent mean ± SD. **P < .01 compared with WT mice with FXII knockdown given saline; ###P < .001 compared with HRG−/− mice with FXII knockdown given saline (analysis of variance, Holm-Šídák method). NS, not significant.
HRG is localized with platelets and fibrin in pulmonary thrombi induced by polyP
Immunohistochemical analysis was used to assess the distribution of HRG, platelets, and fibrin in pulmonary thrombi induced by polyP administration. We have previously shown that HRG binds to fibrin and that HRG localizes with platelets and fibrin in the arterial thrombi induced by FeCl3 application.18 In wild-type mice given polyP, HRG localized with platelets and fibrin in the pulmonary thrombi (Figure 6A-C). HRG was also found on the endothelium (Figure 6A-B), confirming a previous report that HRG is found in endothelial cells.27 As expected, no HRG was detected in the thrombi in HRG−/− mice (Figure 6D-E). These results confirm that HRG localizes with platelets and fibrin in the thrombi induced by polyP administration.

Localization of HRG with platelets and fibrin in pulmonary thrombi. Frozen lung sections harvested from wild-type (WT; panels A-C) or HRG−/− (panels D-E) mice 60 minutes after IP polyP administration were subjected to immunofluorescence using antibodies against HRG, fibrin, and platelets. 4′,6-Diamidino-2-phenylindole (DAPI) was used as a nuclear stain. The combinations for immunostaining were paired as follows: panels A and D, HRG (green) and fibrin/fibrinogen (red); panels B and E, platelets (green) and HRG (red); and panel C, platelets (green) and fibrin/fibrinogen (red). Sections were visualized by using a ×100 objective lens and an Olympus BX41 microscope equipped with a DP72 camera (Olympus, Tokyo, Japan). Scale bars represent 100 μm.
Localization of HRG with platelets and fibrin in pulmonary thrombi. Frozen lung sections harvested from wild-type (WT; panels A-C) or HRG−/− (panels D-E) mice 60 minutes after IP polyP administration were subjected to immunofluorescence using antibodies against HRG, fibrin, and platelets. 4′,6-Diamidino-2-phenylindole (DAPI) was used as a nuclear stain. The combinations for immunostaining were paired as follows: panels A and D, HRG (green) and fibrin/fibrinogen (red); panels B and E, platelets (green) and HRG (red); and panel C, platelets (green) and fibrin/fibrinogen (red). Sections were visualized by using a ×100 objective lens and an Olympus BX41 microscope equipped with a DP72 camera (Olympus, Tokyo, Japan). Scale bars represent 100 μm.
Discussion
Studies have highlighted the importance of the contact system in the pathogenesis of thrombosis and identify naturally occurring polyanions, such as polyP, RNA, and DNA, as initiators of this pathway.5,8,18 Because the contact system is initiated by the activation of FXII, it is critical to understand how FXIIa is regulated in vivo.2,28,29 We have previously shown that HRG binds FXIIa with high affinity, thereby inhibiting FXII autoactivation and attenuating downstream activation of coagulation.17,18 HRG also binds DNA and RNA and attenuates their procoagulant activity.18,30 Together, these results suggest that HRG serves as a natural regulator of the contact system. The current study extends these findings by showing that: (1) HRG binds polyP with high affinity and attenuates its procoagulant activity; (2) the procoagulant effect of polyP is greater in HRG-depleted human plasma than in HRG-sufficient plasma; (3) the procoagulant and prothrombotic effects of polyP are greater in HRG−/− mice than in wild-type mice; and (4) FXII knockdown abrogates the procoagulant and prothrombotic effects of polyP in both HRG−/− and wild-type mice. Therefore, the procoagulant and prothrombotic effects of polyP are FXII dependent and modulated by HRG.
We used short-chain polyP consisting of ∼70 phosphate units in these studies to mimic the polyP released from activated platelets. This polyP preparation promotes FXII activation in a purified system and accelerated plasma clotting via the contact system because it shortened the aPTT but not the prothrombin time. The capacity of short-chain polyP to promote clotting is consistent with some studies4,10,31,32 but at odds with others which suggest that only longer chain polyP promotes FXII activation.11,33,34 Gel analysis confirmed the short nature of this polyP preparation. The procoagulant and prothrombotic effects of short-chain polyP in mice are lost when FXII is knocked down. Therefore, polyP composed of ∼70 phosphate units promotes clotting by activating FXII, an effect attenuated by HRG.
Previous studies have shown that polyP induces thrombosis in mice when administered intravenously in high doses.10,23,33,35,36 With such administration, mice die in <5 minutes and have extensive pulmonary thrombosis.10,23,33 To enable comparison of the effects of polyP in HRG−/− and wild-type mice, we developed a nonsurgical and nonlethal thrombosis model wherein polyP is administrated intraperitoneally. When administered in this manner, polyP is absorbed in a time-dependent manner, as evidenced by progressive shortening of recalcification times and the aPTT, and progressive enhancement of thrombin generation. Consistent with its capacity to modulate polyP activity, functional plasma polyP concentrations were twofold higher in HRG−/− mice than in wild-type mice. This model therefore enabled identification of HRG as a modulator of the procoagulant and prothrombotic effects of polyP.
HRG is a multidomain protein that interacts with a diverse group of ligands, including plasminogen, fibrinogen, and FXIIa.12 HRG attenuates the procoagulant and prothrombotic effects of polyP by binding it in a zinc-dependent manner. Such binding is consistent with the capacity of polyP to bind other polyanions, such as heparin, DNA, and RNA.18,24 The affinity of HRG for polyP (Kd value of 152 ± 9 nM) is lower than that for DNA and RNA, which both bind HRG with Kd values of 1.3 ± 0.8 nM.18,30 Nonetheless, with the plasma concentration of HRG ranging from 1 to 3 µM, polyP released from activated platelets is likely to be bound to HRG. As with polyP, HRG is found in platelets and is released upon platelet activation.37,38 Consistent with this concept, we show the localization of HRG with platelets and fibrin in pulmonary thrombi induced by polyP administration. HRG also is found on the endothelium. Therefore, HRG is ideally positioned to downregulate the procoagulant and prothrombotic effects of polyP released from activated platelets at sites of vascular injury.
FXII was knocked down by using an ASO to confirm the role of the contact system in our polyP-induced thrombosis model. The procoagulant and prothrombotic effects of polyP were abolished with FXII knockdown, a finding consistent with the observation that intravenous polyP has little prothrombotic activity in FXII-deficient mice.10 These results point to activation of FXII as the predominant mechanism by which polyP promotes thrombosis in vivo despite reports of other effects of polyP on coagulation, including enhancement of factor XI and factor V activation by thrombin, altered fibrin polymerization, and attenuation of fibrinolysis.26,31 More studies are needed to determine the contribution of these alternative effects of polyP to thrombosis in settings in which the stimulus to clotting is less robust.
We now have evidence that HRG attenuates thrombosis induced by polyP as well as that induced by vascular injury with FeCl3.18 These findings are consistent with reports that congenital HRG deficiency is associated with thrombophilia.39,40 Furthermore, as a negative acute-phase protein, the levels of HRG decrease with inflammation, infection, and pregnancy, which could contribute to the increased risk of thrombosis observed with these conditions.12,41 In the presence of divalent cations, polyP aggregates into nanoparticles that deposit on the surface of platelets and activate FXII.42,43 Because HRG also is believed to be released by platelets,37 it is well situated to attenuate the platelet-bound polyP. When bound to anionic surfaces, FXIIa is protected from inhibition by C1 inhibitor.44 Consequently, HRG may be a more important regulator than C1 inhibitor when FXIIa is generated on the surface of activated platelets, which could contribute to wound healing as well as hemostasis.45
Our results suggest that HRG serves as a brake for the intrinsic pathway of coagulation via 2 mechanisms. First, HRG neutralizes the procoagulant activity of polyP; second, HRG inhibits the activity of FXIIa. The finding that these two mechanisms operate independently is evidenced by the fact that HRG attenuates silica-induced FXII autoactivation even though HRG does not bind silica.17 Additional studies are needed to parse out the relative contribution of these 2 mechanisms of the inhibitory effects of HRG on the intrinsic pathway.
In summary, we show that polyP-induced thrombosis is enhanced in mice deficient in HRG, an effect that is FXII dependent. Therefore, HRG acts as a molecular brake for the contact system by limiting the procoagulant activity of FXIIa regardless of whether FXII is activated by polyanions such as polyP or by vascular injury.18 With evidence that HRG and FXII are equally important in thrombosis, both may serve as suitable targets for modulation of coagulation.
Acknowledgments
J.I.W. holds the Heart and Stroke Foundation/J. Fraser Mustard Chair in Cardiovascular Research and the Canada Research Chair (Tier 1) in Thrombosis. This work was supported, in part, by grants-in-aid from Canadian Institutes of Health Research (FDN-159928) and the Heart and Stroke Foundation of Canada (G-16-00013163).
Authorship
Contribution: R.A.M. designed and performed experiments, analyzed data, and wrote the paper; J.Z. designed and performed experiments, and analyzed data; T.K.T. performed experiments and produced the visual abstract; J.R.C. and A.S.R. provided vital reagents; and J.C.F. and J.I.W. designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: J.R.C. and A.S.R. are employees of Ionis Pharmaceuticals. J.I.W. has served as a consultant and has received honoraria from Ionis Pharmaceuticals. The other authors declare no competing financial interests.
Correspondence: Jeffrey I. Weitz, Thrombosis and Atherosclerosis Research Institute, 237 Barton St E, Hamilton, ON L8L 2X2, Canada; e-mail: weitzj@taari.ca.

