

Key Points
PTPN11 mutations are recurrent alterations in patients with AML and are an independent prognostic factor for poor survival.
The deleterious effect is confined mostly to patients within the ELN favorable risk group and subclonal constellations of PTPN11 mutations.

Abstract
The tyrosine-protein phosphatase nonreceptor type 11 (PTPN11) is an important regulator of RAS signaling and frequently affected by mutations in patients with acute myeloid leukemia (AML). Despite the relevance for leukemogenesis and as a potential therapeutic target, the prognostic role is controversial. To investigate the prognostic impact of PTPN11 mutations, we analyzed 1529 adult AML patients using next-generation sequencing. PTPN11 mutations were detected in 106 of 1529 (6.93%) patients (median VAF: 24%) in dominant (36%) and subclonal (64%) configuration. Patients with PTPN11 mutations were associated with concomitant mutations in NPM1 (63%), DNMT3A (37%), and NRAS (21%) and had a higher rate of European LeukemiaNet (ELN) favorable cytogenetics (57.8% vs 39.1%; P < .001) and higher white blood cell counts (P = .007) compared with PTPN11 wild-type patients. In a multivariable analysis, PTPN11 mutations were independently associated with poor overall survival (hazard ratio [HR]: 1.75; P < .001), relapse-free survival (HR: 1.52; P = .013), and a lower rate of complete remission (odds ratio: 0.46; P = .008). Importantly, the deleterious effect of PTPN11 mutations was confined predominantly to the ELN favorable-risk group and patients with subclonal PTPN11 mutations (HR: 2.28; P < .001) but not found with dominant PTPN11 mutations (HR: 1.07; P = .775), presumably because of significant differences within the rate and spectrum of associated comutations. In conclusion, our data suggest an overall poor prognostic impact of PTPN11 mutations in AML, which is significantly modified by the underlying cytogenetics and the clonal context in which they occur.
Introduction
Acute myeloid leukemia (AML) is a clonal hematologic malignancy and is heterogeneous with respect to clinical presentation, outcome, and the underlying molecular landscape.1 To define clinically relevant disease subentities, several recurrent genetic abnormalities are included in current molecular risk categories summarized in the 2016 World Health Organization2 classification and the 2017 European LeukemiaNet (ELN) recommendations for diagnosis and management of AML.3 Beside specific chromosomal aberrations such as t(15;17), t(8;21), and inv(16), molecular groups incorporate gene mutations in NPM1, CEBPAbiallelic, and provisionally, RUNX1 as full entities.2,3 Moreover, the integration of sequencing data from large-scale genomic studies in patients with AML identified a series of frequently mutated genes, functionally linked to epigenetic modification (eg, ASXL1, DNMT3A, IDH1/2, TET2), apoptosis (TP53), or cell signaling (FLT3ITD, NRAS, KIT), which are significant for prognosis and treatment.1-4
In addition to established prognostic markers, the tyrosine-protein phosphatase nonreceptor type 11 (PTPN11) is frequently affected by somatically acquired mutations in AML.1,5-9 PTPN11 comprises 2 N-terminal Src homology 2 (SH2) domains, a protein tyrosine phosphatase (PTP) catalytic domain, and a COOH terminus.10 The gene encodes for the ubiquitously expressed cytoplasmic phosphatase SHP2, which has a significant role for cell growth and differentiation by mediating cellular response to hormones and cytokines.10,11 In normal hematopoiesis, PTPN11 is a positive (signal-enhancing) component downstream of cell surface growth factor receptors and acts as an important regulator of RAS/MAPK signaling pathways.10,11 In leukemogenesis, somatic gain-of-function mutations located at the N-SH2/PTP domains of PTPN11 block autoregulation of SHP2 catalytic activity. As a consequence, upregulation of SHP2 was shown to induce hypersensitivity to granulocyte-macrophage colony-stimulating factor and hyperactivation of the RAS signaling axis in hematopoietic progenitor cells, resulting in leukemic transformation.12-15
Beside adult AML, PTPN11 mutations have been implicated in a variety of hematologic malignancies, including juvenile myelomonocytic leukemia, childhood AML, myelodysplastic syndrome, and B-cell acute lymphoblastic leukemia.5,7,9 Moreover, mutations of PTPN11 were detected in diverse solid cancer types (eg, lung, breast, colon cancer, and neuroblastoma)16 and are associated with developmental pathologies such as Noonan syndrome.17 Depending on the type of cancer, PTPN11 was identified as tumor suppressor or proto-oncogene, which points at a mutual role of PTPN11 for malignant progression18-20 and indicates the potential of SHP2 as therapeutic target.21-24
More recently, the detection of PTPN11 mutations in adult patients with AML was associated with certain clinical features (ie, high rate of concomitant NPM1 mutations) and adverse prognosis (shorter overall survival [OS]).25-27 In contrast, other recent reports did not observe a significant effect of PTPN11 mutations on outcome in AML28,29 or, vice versa, an improved prognosis from the association with favorable genetic characteristics.30 Hence, the clinical relevance of PTPN11 mutations in AML still remains controversial and needs further elucidation.
To investigate the prevalence and prognostic impact of PTPN11 mutations in adult patients with AML, we analyzed a large cohort of 1529 patients with newly diagnosed AML. Using next-generation sequencing, we identified an overall prevalence of ∼7% PTPN11 mutations in adult AML, with distinct molecular and clinical features. In a multivariable analysis, PTPN11 mutations were an independent prognostic factor for worse outcome, mainly associated with a significant deleterious effect in the ELN favorable risk group. Most importantly, we found a profound and not previously reported association of the clonal rank (dominant vs subclonal) of PTPN11 mutations with the comutational spectrum and clinical outcome, which might help to further refine prognostic implications of PTPN11 mutations in adult AML.
Methods
Patients
We retrospectively screened 1529 adult patients with newly diagnosed AML for the detection of mutations in PTPN11. Patients were included if they had available material (genomic DNA) at diagnosis. All patient samples were obtained and analyzed with written informed consent of the patients. The study was in agreement with the Helsinki declaration and approved by the ethical board of the Technical University Dresden (EK98032010). The protocols were registered for prospective trials of the Study Alliance Leukemia (SAL) with NCT numbers 00180115 (AML96), 00180102 (AML2003), 00180167 (AML60+), and 00893373 (SORAML). Detailed descriptions of the treatment protocols have been published previously;31-34 all protocols included intensive induction chemotherapy and consolidation treatment. AML was defined as de novo when no previous malignancy and no previous treatment were reported. AML was defined as secondary (sAML) or therapy-associated AML (tAML) when myeloid neoplasms or exposure to chemo- and/or radiotherapy were documented before AML diagnosis. Early death was defined as death of any cause within 30 days after initial diagnosis (ED30). Remission and survival criteria were defined according to ELN2017 recommendations.3 Event-free survival (EFS) was defined as the time from diagnosis to death (from any cause), relapse, or failure to achieve a complete remission (CR) after induction.
Patient samples
All molecular studies were performed on DNA isolated from bone marrow aspirates or peripheral blood taken at diagnosis. Genomic DNA from samples was extracted using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany) and quantified with the NanoDrop spectrophotometer. All samples were collected with written informed consent and after approval by the local ethics committee of the Medical Faculty Carl Gustav Carus Dresden, Germany.
Molecular analysis
Profiling of PTPN11 mutational status and associated comutations was done by targeted resequencing using the TruSight Myeloid assay (Illumina, San Diego, CA), according to manufacturer’s recommendations and as described previously.35 The panel targets 54 genes associated with myeloid neoplasms. For PTPN11, the panel covers all relevant mutational hotspots of the N-terminal SH2 domain (exon 3) and the PTP domain (exon 13) of PTPN11. Briefly, for each reaction, 50 ng genomic DNA was used. Library preparation was done as recommended by the manufacturer (TruSight Myeloid Sequencing Panel Reference Guide 15054779 v02, Illumina). Samples were sequenced paired-end on a NextSeq (150-bp Paired End) or MiSeq (300-bp Paired End) next-generation sequencing instrument (Illumina). Sequence data alignment of demultiplexed FastQ files, variant calling and filtering was done using the Sequence Pilot software package (JSI Medical Systems GmbH, Ettenheim, Germany) with default settings and a 5% variant allele frequency (VAF; dividing the mutant allele by the total alleles present; given in percent) mutation calling cutoff. Human genome build HG19 was used as reference genome for mapping algorithms. PolyPhen scores to predict effect of PTPN11 mutations on amino acid exchange were analyzed using the SeattleSeq annotation pipeline (https://snp.gs.washington.edu/SeattleSeqAnnotation154/). Dichotomization of dominant and subclonal (or secondary) PTPN11 mutations was performed by comparing VAFs of detected PTPN11 mutations with VAFs of comutated genes. For resolution of putative subclonal PTPN11 mutations a minimum difference of ≥10% VAF, compared with the most prominent comutation, was applied. PTPN11 mutations with VAFs higher or similar (<10% difference) to detected comutations were classified dominant. Mutational analysis for FLT3-ITD mutations was done as reported previously.36
Statistical analysis
Categorical variables between groups were compared using the χ2 test or a 2-sided Fisher’s exact test. For continuous variables, the nonparametric Mann-Whitney U test was applied. P < .05 was considered significant. Univariate analyses for the effect of PTPN11 mutations on CR rates were performed using the χ2 test. To evaluate EFS, relapse-free survival (RFS), and OS, the Kaplan-Meier method and log-rank test were used. For multivariable analysis of prognostic factors, Cox-proportional hazard regression models were used for survival end points, and logistic regression models were used for CR. All statistical analyses were performed using the R environment for statistical computing version 4.0.3.
Results
Characterization of PTPN11 mutations and associated comutations
PTPN11 mutations were found in 106 of 1529 (6.9%) patients. A detailed list of all PTPN11 variants is provided in supplemental Table 1. Most patients (n = 97) carried a single mutation in PTPN11, and 9 patients harbored 2 different PTPN11 variants (double mutated). Mutations exclusively comprised missense single nucleotide variants and were detected predominantly (76%) in the highly conserved N-terminal SH2 domain (24% in PTP domain), relevant for the catalytic function of SHP2 (Figure 1A). PolyPhen score was >0.85 for 94% of variants, predicting a damaging impact on amino acid substitution for most mutations (supplemental Table 1). The most common alterations were A72T and A72V (found in 12 patients each). Other recurrent pathogenic single nucleotide variants, previously associated with rasopathy, Noonan syndrome, and AML, were detected at residues G60, D61, E69, F71, and E76 (N-SH2), as well as S503, G503, and T507 (PTP) (Figure 1A).
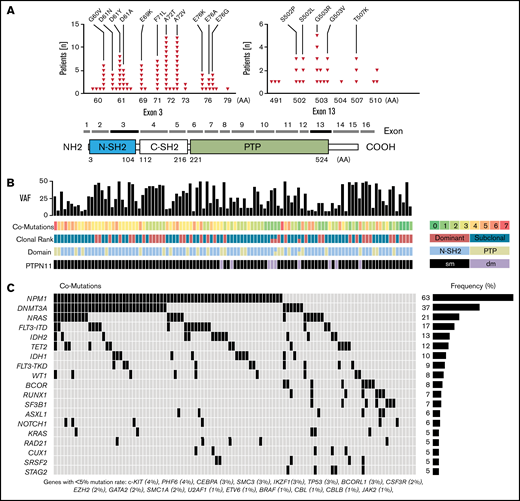
PTPN11 mutations and associated comutations. (A) Schematic illustration showing the position of acquired PTPN11 mutations in 106 AML patients and the domain structure of PTPN11: 2 Src homology 2 domains (N-SH2 and C-SH2) and a PTP (protein tyrosine phosphatase) domain. Recurrent alterations and their frequency in AML patients are indicated for PTPN11 exons 3 and 13. (B) PTPN11 VAFs, number of comutations (coded by color), PTPN11 clonal rank (dominant vs subclonal), affected functional domains (N-SH2 vs PTP), and the number of PTPN11 single (sm) or double mutated (dm) patients. (C) Associated comutations in PTPN11-mut patients.
PTPN11 mutations and associated comutations. (A) Schematic illustration showing the position of acquired PTPN11 mutations in 106 AML patients and the domain structure of PTPN11: 2 Src homology 2 domains (N-SH2 and C-SH2) and a PTP (protein tyrosine phosphatase) domain. Recurrent alterations and their frequency in AML patients are indicated for PTPN11 exons 3 and 13. (B) PTPN11 VAFs, number of comutations (coded by color), PTPN11 clonal rank (dominant vs subclonal), affected functional domains (N-SH2 vs PTP), and the number of PTPN11 single (sm) or double mutated (dm) patients. (C) Associated comutations in PTPN11-mut patients.
Mutations were detected with a median VAF of 24% (range, 5%-52%) in clonal and subclonal configuration (Figure 1B). Most mutations were found at subclonal levels (64%; PTPN11SUB) with significantly lower allele frequency (>10% difference of VAFs) compared with comutated genes (Figure 1B). Dominant PTPN11 variants (PTPN11DOM) with VAFs higher or similar to associated comutations were detected in 38 patients (36%). PTPN11 mutations were significantly associated with concomitant mutations in NPM1 (63% vs 30%; P < .001), and there was a trend for higher rates of DNMT3A (37% vs 28%; P = .056) and NRAS (21% vs 16%; P = .196) mutations (mut) compared with PTPN11 wild-type (wt) patients (Figure 1C; Table 1). No major difference between PTPN11-mut and -wt patients was detected for other frequently mutated genes in AML, for example, FLT3ITD (17%), FLT3TKD (9%), IDH1/2 (10/13%), and TET2 (12%).
Clinical and molecular characteristics of PTPN11-mut and PTPN11-wt patients
| Parameter | PTPN11-wt | PTPN11-mut | P |
|---|---|---|---|
| No of patients (n) | 1423 | 106 | |
| Age (y), median (IQR) | 55 (44-64.5) | 54 (42-63) | 0.377 |
| Sex, n (%) | .857 | ||
| Female | 678 (47.6) | 52 (49.1) | |
| Male | 745 (52.4) | 54 (50.9) | |
| Disease status, n (%) | .313 | ||
| De novo | 1198 (85.1) | 89 (85.6) | |
| tAML | 159 (11.3) | 14 (13.5) | |
| sAML | 50 (3.6) | 1 (1) | |
| ELN risk 2017, n (%) | <.001 | ||
| Favorable | 511 (39.1) | 59 (57.8) | |
| Intermediate | 543 (41.5) | 23 (22.5) | |
| Adverse | 253 (19.4) | 20 (19.6) | |
| Complex aberrant karyotype, n (%) | .154 | ||
| No | 1139 (87.5) | 92 (92.9) | |
| Yes | 162 (12.5) | 7 (7.1) | |
| FLT3-ITD, n (%) | .200 | ||
| No | 1089 (77.1) | 88 (83) | |
| Yes | 323 (22.9) | 18 (17) | |
| NPM1 mutation, n (%) | <.001 | ||
| No | 984 (70.3) | 38 (36.5) | |
| Yes | 416 (29.7) | 66 (63.5) | |
| ECOG score, n (%) | .064 | ||
| 0-1 | 842 (71.7) | 53 (61.6) | |
| 2-4 | 333 (28.3) | 33 (38.4) | |
| Laboratory, median (IQR) | |||
| WBC (Gpt/L) | 18.8 (4.4-54.37) | 26.76 (13.5-68.17) | .007 |
| Hemoglobin (mmol/L) | 5.9 (5.03-7.01) | 5.77 (5.03-6.77) | .408 |
| Platelets (Gpt/L) | 50 (27-94) | 54 (32-83) | .585 |
| Peripheral blasts (%) | 42 (12-75) | 38 (13.5-64) | .477 |
| LDH (U/L) | 448 (280-786) | 473.5 (374.25-853.75) | .025 |
| BM blasts (%) | 63 (44.38-79) | 62.75 (45.5-78) | .708 |
| Clinical outcome | |||
| CR rate, n (%) | 1038 (72.9) | 71 (67) | .008* |
| ED30, n (%) | 80 (5.6) | 12 (11.3) | .001* |
| EFS (mo), median (95% CI) | 7.42 (6.7-8.3) | 7.53 (4.41-10.91) | .022* |
| RFS (mo), median (95% CI) | 18.41 (15.87-23.34) | 14.66 (8.02-28.37) | .013* |
| OS (mo), median (95% CI) | 19.23 (17.16-21.76) | 13.44 (10.13-20.25) | <.001* |
| Parameter | PTPN11-wt | PTPN11-mut | P |
|---|---|---|---|
| No of patients (n) | 1423 | 106 | |
| Age (y), median (IQR) | 55 (44-64.5) | 54 (42-63) | 0.377 |
| Sex, n (%) | .857 | ||
| Female | 678 (47.6) | 52 (49.1) | |
| Male | 745 (52.4) | 54 (50.9) | |
| Disease status, n (%) | .313 | ||
| De novo | 1198 (85.1) | 89 (85.6) | |
| tAML | 159 (11.3) | 14 (13.5) | |
| sAML | 50 (3.6) | 1 (1) | |
| ELN risk 2017, n (%) | <.001 | ||
| Favorable | 511 (39.1) | 59 (57.8) | |
| Intermediate | 543 (41.5) | 23 (22.5) | |
| Adverse | 253 (19.4) | 20 (19.6) | |
| Complex aberrant karyotype, n (%) | .154 | ||
| No | 1139 (87.5) | 92 (92.9) | |
| Yes | 162 (12.5) | 7 (7.1) | |
| FLT3-ITD, n (%) | .200 | ||
| No | 1089 (77.1) | 88 (83) | |
| Yes | 323 (22.9) | 18 (17) | |
| NPM1 mutation, n (%) | <.001 | ||
| No | 984 (70.3) | 38 (36.5) | |
| Yes | 416 (29.7) | 66 (63.5) | |
| ECOG score, n (%) | .064 | ||
| 0-1 | 842 (71.7) | 53 (61.6) | |
| 2-4 | 333 (28.3) | 33 (38.4) | |
| Laboratory, median (IQR) | |||
| WBC (Gpt/L) | 18.8 (4.4-54.37) | 26.76 (13.5-68.17) | .007 |
| Hemoglobin (mmol/L) | 5.9 (5.03-7.01) | 5.77 (5.03-6.77) | .408 |
| Platelets (Gpt/L) | 50 (27-94) | 54 (32-83) | .585 |
| Peripheral blasts (%) | 42 (12-75) | 38 (13.5-64) | .477 |
| LDH (U/L) | 448 (280-786) | 473.5 (374.25-853.75) | .025 |
| BM blasts (%) | 63 (44.38-79) | 62.75 (45.5-78) | .708 |
| Clinical outcome | |||
| CR rate, n (%) | 1038 (72.9) | 71 (67) | .008* |
| ED30, n (%) | 80 (5.6) | 12 (11.3) | .001* |
| EFS (mo), median (95% CI) | 7.42 (6.7-8.3) | 7.53 (4.41-10.91) | .022* |
| RFS (mo), median (95% CI) | 18.41 (15.87-23.34) | 14.66 (8.02-28.37) | .013* |
| OS (mo), median (95% CI) | 19.23 (17.16-21.76) | 13.44 (10.13-20.25) | <.001* |
BM, bone marrow; CI, confidence interval; CR, complete remission; ED30, early death within 30 days; EFS, event-free survival; ELN, European LeukemiaNET; IQR, interquartile range; LDH, lactate dehydrogenase; OS, overall survival; PB, peripheral blood; RFS, relapse-free survival; sAML, secondary acute myeloid leukemia; tAML, therapy-related acute myeloid leukemia; WBC, white blood cells.
Results of the multivariable regression model.
Associations of PTPN11 mutations with clinical features and outcome
Descriptive statistics of clinical parameters revealed significant differences between PTPN11-mut and -wt patients (Table 1). Patients with PTPN11 mutations were diagnosed significantly more often with favorable cytogenetics (57.8%) according to the ELN-2017 risk criteria compared with PTPN11-wt patients (39.1%; P < .001). Accordingly, PTPN11-mut patients had a lower rate of ELN-2017 intermediate risk (22.5% vs 41.5% in PTPN11-wt patients). No differences were detected for the relative proportion of ELN-2017 adverse risk between both groups (19.4%-19.6%). In addition, PTPN11-mut patients had significantly (P = .007) higher white blood cell (WBC) counts (median 26.7 vs 18.8 Gpt/L; P = .007) and higher concentrations of lactate dehydrogenase (LDH; median 473.5 vs 448 U/L; P = .025) compared with PTPN11-wt patients. Moreover, there was a trend for a higher prevalence of inferior Eastern Cooperative Oncology Group (ECOG) performance status (score 2-4) for PTPN11-mut patients (38.4% vs 28.3%; P = .064).
In contrast, no differences were observed for the age at diagnosis (median, 54 vs 55 years), the presence of a complex karyotype (7.1% vs 12.5%), and platelet counts (median, 54 vs 50 Gpt/L) for PTPN11-mut and -wt patients. Similarly, incidences of de novo AML (85.6%), sAML (13.5%), and tAML (1%) observed in PTPN11-mut patients virtually resembled rates of AML types found in PTPN11-wt patients. Also, bone marrow (and blood) blast counts were similar in both groups.
With respect to clinical outcome, PTPN11-mut patients were significantly associated with poor OS (median, 13.44 vs 19.23 months; P = .026; Figure 2A). In univariate analyses, no significant differences between PTPN11-mut and -wt patients were observed for EFS (P = .381; Figure 2B) and RFS (P = .183; Figure 2C). However, the multivariable analysis with clinical variable such as age, cytogenetics, AML type, and ECOG performance status using Cox proportional hazard regression revealed that PTPN11 mutations are an independent prognostic factor for worse OS (hazard ratio [HR], 1.75; 95% confidence interval [CI], 1.34-2.29; P < .001), RFS (HR, 1.52; 95% CI, 1.09-2.12; P = .013), EFS (HR, 1.35; 95% CI, 1.04-1.75; P = .022), and lower rates of complete remission (CR1; odds ratio, 0.46; 95% CI, 0.26-0.82; P = .008; Table 1; Figure 2G). Moreover, PTPN11 mutations were significantly associated with elevated rates of early death (ED30) in univariate (11.3% vs 5.6%; P = .02) and multivariable analyses (HR, 3.61; 95% CI, 1.71-7.59; P = .001; Figure 2G). However, causes of early death were predominantly caused by infectious diseases. The deleterious effect of PTPN11 mutations on outcome was similar for patients with (OS HR, 1.848; 95% CI, 0.726-4.704; P = .198; n = 10) or without (OS HR, 1.798; 95% CI, 1.351-2.393; P < .001; n = 96) allogeneic stem cell transplantation performed in first remission. Also, no differential effect for clinical outcome was detected for the affected functional domains of PTPN11, with a median OS of 12.9 (N-SH2) vs 13.6 months (PTP).
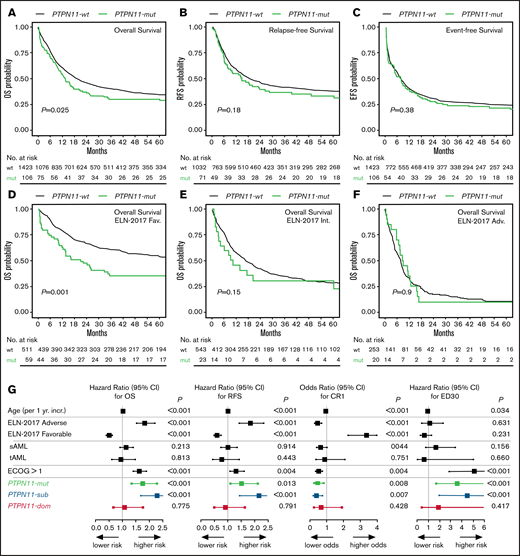
Correlation of PTPN11 mutational status with clinical outcome. Kaplan-Meier analysis showing the probability of (A) OS, RFS, and EFS for AML patients with (mut; green) or without (wt; black) mutation in PTPN11, as well as OS in ELN-2017 favorable (D), intermediate (E), and adverse (F) risk groups. Numbers at risk are presented below each figure. (G) Results of the multivariable analysis for PTPN11-mut (n = 106; green) vs PTPN11SUB (n = 68; blue) vs PTPN11DOM (n = 38; red) mutational status.
Correlation of PTPN11 mutational status with clinical outcome. Kaplan-Meier analysis showing the probability of (A) OS, RFS, and EFS for AML patients with (mut; green) or without (wt; black) mutation in PTPN11, as well as OS in ELN-2017 favorable (D), intermediate (E), and adverse (F) risk groups. Numbers at risk are presented below each figure. (G) Results of the multivariable analysis for PTPN11-mut (n = 106; green) vs PTPN11SUB (n = 68; blue) vs PTPN11DOM (n = 38; red) mutational status.
The comparison of survival times across each ELN risk category demonstrated a deleterious effect of PTPN11 mutations predominantly within the favorable risk group (HR, 1.97; 95% CI, 1.42-2.74; P < .001; Figure 2D). No significant effects of PTPN11 mutations for OS were observed in the intermediate (HR, 1.41; 95% CI, 0.87-2.26; P = .153; Figure 2E) and adverse risk groups (HR, 0.96; 95% CI, 0.59-1.56; P = .897; Figure 2F). Similar associations of poor outcome in PTPN11-mut patients within the favorable ELN group were detected for EFS, CR1, and ED30 (supplemental Table 2).
Impact of PTPN11 clonal rank on clinical outcome
To address the impact of PTPN11 mutations on clinical outcome in more detail and because of the high prevalence of subclonal PTPN11 variants, survival was next analyzed related to the clonal rank (dominant vs subclonal) of detected PTPN11 variants (Figure 3). Compared with patients with dominant PTPN11 mutations (PTPN11DOM), the detection of PTPN11 mutations in subclonal configuration (PTPN11SUB) was associated with poorer median OS (10.38 vs 29.88 months; P = .0018; Figure 3A), RFS (8.62 vs 39.48 months; P = .0061; Figure 3B), and EFS (5.39 vs 11.94 months; P = .13; Figure 3C), as well as higher rates of ED30 (14.7% vs 5.3%; P = .003) and lower rates of CR1 (61.8% vs 76.3%; P = .046). In line, univariate (supplemental Table 3) and multivariable analyses (Figure 2G) to assess the specific effect of PTPN11SUB demonstrated an inferior outcome compared with PTPN11-wt (and PTPN11DOM) patients. In contrast, no significant differences for clinical outcome were observed between PTPN11DOM and PTPN11-wt cases. Similar to the overall effect of PTPN11-mut on survival, PTPN11SUB configuration was associated with poor outcome, in particular within favorable (Figure 3D) and intermediate cytogenetic contexts (Figure 3E), whereas no differences between PTPN11SUB and PTPN11DOM (and PTPN11-wt) were detected in the ELN adverse risk group (Figure 3F).
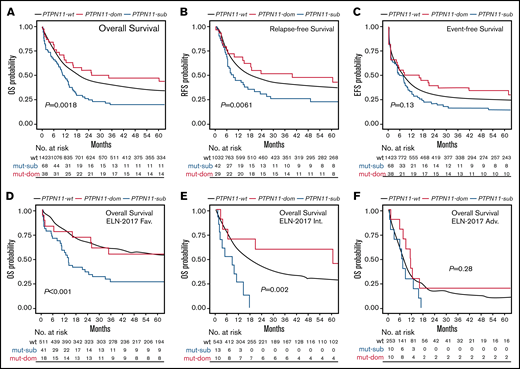
Association of PTPN11 clonal rank with clinical outcome. Kaplan-Meier analysis showing the probability of (A) OS, RFS, and EFS for AML patients without (wt; black) or with PTPN11 mutation in subclonal (PTPN11SUB; blue) or dominant (PTPN11DOM; red) configuration, as well as OS in ELN-2017 favorable (D), intermediate (E), and adverse (F) risk groups. Numbers at risk are presented below each figure.
Association of PTPN11 clonal rank with clinical outcome. Kaplan-Meier analysis showing the probability of (A) OS, RFS, and EFS for AML patients without (wt; black) or with PTPN11 mutation in subclonal (PTPN11SUB; blue) or dominant (PTPN11DOM; red) configuration, as well as OS in ELN-2017 favorable (D), intermediate (E), and adverse (F) risk groups. Numbers at risk are presented below each figure.
In addition to clinical outcome, PTPN11SUB patients were associated with distinct molecular features (Figure 4). PTPN11SUB mutations were detected with significantly lower VAFs (median, 16.5%; interquartile range [IQR], 9%-25.75%; P < .001), compared with PTPN11DOM variants (median, 42.5%; IQR, 34%-48%; Figure 4B) and had a significantly (P < .001) higher rate of associated comutations (median, 3; range, 1-7) compared with PTPN11DOM (median, 2; range, 0-5; Figure 4C). Moreover, the mutational spectrum differed between both groups, with significantly higher rates of concomitant mutations in TET2 (17.6% vs 2.6%; P = .023), NRAS (27.9% vs 7.8%; P = .014), and RUNX1 (10.3% vs 0%; P = .041) for PTPN11SUB vs PTPN11DOM (Figure 4A). Also, molecular lesions in ASXL1 and FLT3TKD were almost exclusively detected within the PTPN11SUB group. No differences were observed for the association with mutations in DNMT3A (36.7% vs 36.8%), NPM1 (66.1% vs 57.9%), and FLT3ITD (16.2% vs 18.4%). A schematic representation of typical clonal configurations for PTPN11DOM and PTPN11SUB AMLs is shown in Figure 4D-E.
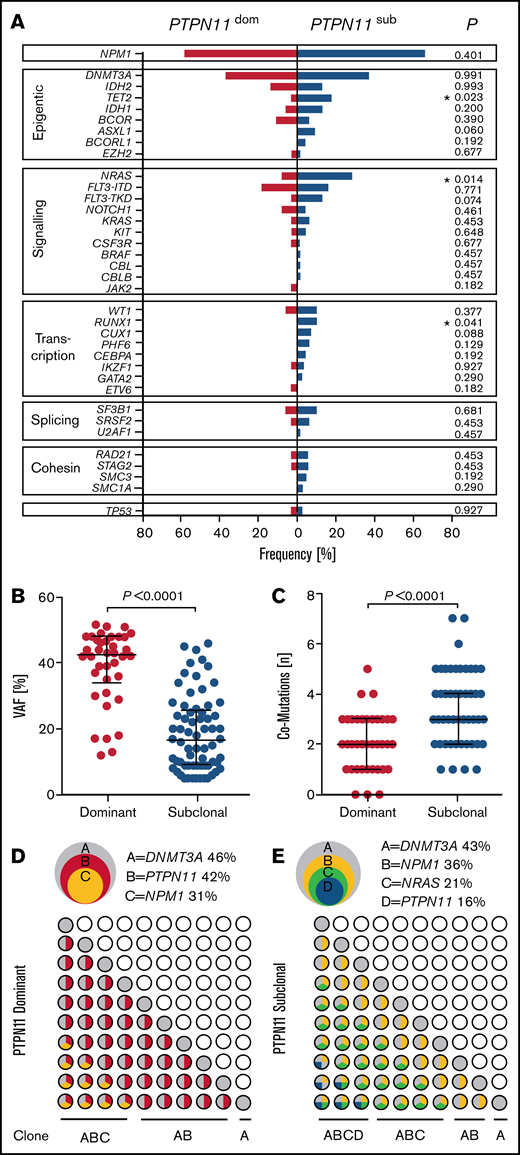
Correlations of PTPN11 clonal hierarchy with molecular features. (A) Rates of associated comutations in patients with PTPN11 mutations in subclonal (PTPN11SUB; blue) and dominant (PTPN11DOM; red) configuration. Significant differences are indicated. (B) VAFs of subclonal (n = 68) and dominant (n = 38) PTPN11 mutations. (C) Overall number of comutations associated with subclonal (n = 68) and dominant (n = 38) PTPN11 mutations. (D-E) Schematic illustration of representative clonal hierarchies (depicted from VAFs of PTPN11 mutations and comutations) in PTPN11DOM (D) and PTPN11SUB (E) AMLs of 2 PTPN11-mut patients (overlapping circles, top) and potential sequential acquisition of PTPN11 mutations (and comutations) as early (PTPN11DOM) or late (PTPN11SUB) event during clonal evolution (grid of circles, bottom).
Correlations of PTPN11 clonal hierarchy with molecular features. (A) Rates of associated comutations in patients with PTPN11 mutations in subclonal (PTPN11SUB; blue) and dominant (PTPN11DOM; red) configuration. Significant differences are indicated. (B) VAFs of subclonal (n = 68) and dominant (n = 38) PTPN11 mutations. (C) Overall number of comutations associated with subclonal (n = 68) and dominant (n = 38) PTPN11 mutations. (D-E) Schematic illustration of representative clonal hierarchies (depicted from VAFs of PTPN11 mutations and comutations) in PTPN11DOM (D) and PTPN11SUB (E) AMLs of 2 PTPN11-mut patients (overlapping circles, top) and potential sequential acquisition of PTPN11 mutations (and comutations) as early (PTPN11DOM) or late (PTPN11SUB) event during clonal evolution (grid of circles, bottom).
With respect to clinical characteristics, no major differences between PTPN11DOM and PTPN11SUB were observed. As an exception, patients with PTPN11SUB were significantly (P = .003) older (median, 56.5 years; IQR, 46.75-68.25 years) compared with PTPN11DOM cases (median, 50.2 years; IQR, 31.5-56.5 years). Also, LDH concentrations (U/L) were significantly (P = .027) elevated in the group of PTPN11DOM (median, 544.5 U/L; IQR, 445.75-957.5 U/L), whereas LDH levels of PTPN11SUB patients (median, 442.7 U/L, IQR, 326.75-780.5 U/L) were comparable to values measured in PTPN11-wt patients.
Discussion
In the present study, we analyzed a large cohort of 1529 adult AML patients for the prevalence of PTPN11 mutations. We confirm that PTPN11 mutations are recurrent alterations in patients with AML, which are independently associated with poor clinical outcome and distinct molecular and clinical features. Importantly, a more detailed analysis of PTPN11 clonal hierarchy and ELN risk categories revealed that the deleterious effect of PTPN11 mutations is mostly confined to subclonal constellations of PTPN11 mutations and patients with a favorable cytogenetic context.
Generally, PTPN11 mutations were detected with a frequency of 7%, which is in the range of prevalences (4%-12%) previously reported for AML.1,25,26,28-30 In line with published data,26,28-30 mutations predominantly affected the N-terminal SH2 domain of PTPN11, involved in basal inhibition of SHP2,12-15 and were found at subclonal levels in most patients.26 This is consistent with a predominance of RAS signaling gene mutations (eg, FLT3TKD, NRAS, and KIT) as secondary or subclonal lesions in AML that ether emerge or are lost during clonal disease evolution.1,37,38 The importance of individual PTPN11 variants (eg, E69K, G503A, and E76K), recurrently detected in our cohort, for RAS signaling hyperactivation, leukemic transformation, and chemoresistance in AML, has been summarized recently.26
Similar to previous findings on clinical phenotype in AML, PTPN11-mut patients had significantly higher WBC counts26,30 and were associated with a characteristic mutational spectrum (ie, NPM1 and NRAS) frequently found comutated in PTPN11-mut AML,25,26,28-30 which is suggestive for a distinct pathway of pathogenesis. Interestingly, the association with NRAS mutations in cases with dominant PTPN11 mutations contradicts a presumed mutual exclusivity of activating mutations in PTPN11 (or FLT3 and KIT) with downstream RAS mutations (NRAS/KRAS) initially observed in hematologic malignancies.38-40
However, except for general similarities with respect to PTPN11 prevalences and some phenotypic and molecular features, no consensus has been reached for prognostic implications of PTPN11-mut AML in the literature.25-30 In part, this might be attributed to considerable differences in patients’ characteristics (and numbers) of the analyzed cohorts. For example, in our study, PTPN11-mut patients largely (57.8%) clustered within the ELN 2017 favorable risk category, mainly because of a high rate of associated NPM1 mutations (63%), which confirms recent reports on rates of NPM1 mutations (60.5%-61%) and the underlying cytogenetics in PTPN11-mut AML.29,30 This is in conflict with other documentations, with a low rate (18%)26 or absence (0%)27 of favorable cytogenetics in PTPN11-mut AML, likely because of a lower frequency of concomitant NPM1 mutations (29% and 22%).26,27 In addition to the cytogenetic context, previous work on PTPN11-mut AML varied considerably with respect to the age of PTPN11-mut patients at diagnosis (eg, 67-7025-27 vs <60 years29,30) and the respective treatment regimes (eg, high vs low intensity).
In our large cohort of newly diagnosed and intensively treated AML patients, the median age at diagnosis was 55 years (54 years for PTPN11-mut patients). For this clinically relevant setting, multivariable analysis revealed that PTPN11 mutations are an independent prognostic factor for worse outcome. In addition, we show that the overall unfavorable prognostic effect is mainly substantiated by an adverse impact of PTPN11-mut in the ELN 2017 favorable (and to some extent in the intermediate) risk group, whereas no such association was observed in patients with adverse cytogenetics. This is in disagreement with recent published data for AML patients (n = 410) treated with high-intensity therapy, where PTPN11 mutations were associated with poor survival, mainly in the ELN intermediate and adverse groups.26
The latter work also reported on an (insignificant) effect of VAFs > 40% for worse outcome. Here, we could not confirm an association of high allelic fraction PTPN11 variants with poor survival. In contrast, we observed an inferior impact on outcome for PTPN11 mutations in subclonal configuration (PTPN11SUB), which were associated with significantly lower allelic fraction, higher rates of comutations, and different comutational patterns (ie, NRAS and TET2) compared with AMLs with dominant PTPN11 clones, suggesting a potential impact of cooperating mutant proteins. TET2 mutations were recently shown to confer a poor outcome in patients with myelodysplastic syndrome and concomitant secondary SF3B1 mutations, whereas no adverse effects of TET2 mutations were detected for the association with dominant SF3B1 variants in the same cohort.41 Similar associations of distinct dominant/subclonal gene–gene interactions with specific clinical features were reported previously and are especially pronounced for NPM1-mutated AML.1,42 A higher overall rate of associated comutations in PTPN11SUB patients (compared with PTPN11DOM) may also coincide with a higher clonal diversity and the presence of various treatment escape mechanisms because of the deregulation of multiple cell signaling pathways and a higher degree of genomic instability.1,37,38 If confirmed, these results would suggest that the acquisition of a subclonal mutation as a late event, occurring with multiple other adverse features, may lead to poor outcome. In contrast, when PTPN11 was a dominant driver mutation, clinical outcomes were similar (or even more favorable) compared with PTPN11-wt patients, indicating a less aggressive disease in these patients. Thus, considering these substantial differences, the overall deleterious effect of PTPN11 mutations in AML might be an epiphenomenon, depending on the clonal constellation in which they occur. The impact of this effect is presumably diminished in PTPN11-mut AML with ELN adverse cytogenetics, where no prognostic difference between PTPN11SUB and PTPN11DOM patients was detected. However, to fully understand the prognostic role of PTPN11 clonal hierarchy and to discriminate between true subclonal and secondary PTPN11 mutations, future investigations may track clonal progression at disease recurrence in PTPN11-mut AML. Furthermore, single-cell sequencing will potentially provide a higher resolution to address the dynamics of individual clones present in the same pathway (eg, PTPN11 and NRAS) and their impact for relapse development.43
Taken together, our data suggest that PTPN11 mutations are recurrent alterations in patients with AML and are an independent prognostic factor for poor survival, with worse clinical outcome in patients within the ELN favorable risk group. The association with distinct clinical and molecular features, points to a specific genesis in this subset of AML. Importantly, the deleterious outcome in PTPN11-mut patients was found to be confined to subclonal configurations of PTPN11 mutations, which are associated with a higher rate of concomitant mutations and a different mutational spectrum, as compared with PTPN11 variants in dominant clonal rank.
Acknowledgments
The authors thank M. Böhm and M. Hartwig for skillful technical assistance.
Authorship
Contribution: C.T., M.M., J-N.E., and S.S. conceived the work, acquired and analyzed the data, and interpreted the data; S.S. performed bioinformatic analysis and drafted the manuscript; M.K. provided statistical analysis; G.E. and M.B. provided administrative support; and all authors provided samples, collected data, and have seen and approved the manuscript.
Conflict-of-interest disclosure: C.T. is chief executive officer and co-owner of AgenDix GmbH, a company performing molecular diagnostics. The remaining authors declare no competing financial interests.
Correspondence: Christian Thiede, Universitätsklinikum Carl Gustav Carus, Medizinische Klinik und Poliklinik I, Fetscherstr 74, 01307 Dresden, Germany; e-mail: christian.thiede@uniklinikum-dresden.de.
Appendix: participating centers of the Study Alliance Leukemia (SAL)
Universitätsklinikum Aachen der RWTH, Aachen, Tim H. Brümmendorf; Klinikum Altenburger Land GmbH, Altenburg, Armin Schulz-Abelius; Klinikum Augsburg, Augsburg, Martin Trepel; Sozialstiftung Bamberg, Bamberg, Martina Teichmann; Klinikum Bayreuth GmbH, Bayreuth, Alexander Kiani; Helios Klinikum Berlin-Buch, Berlin, Bertram Glaß; Städt. Kliniken Bielefeld gGmbH, Bielefeld, Martin Görner; Augusta-Kranken-Anstalt gGmbH, Bochum, Dirk Behringer; Ev. Diakonie-Krankenhaus GmbH, Bremen, Johannes Kullmer; Klinikum Chemnitz GmbH, Chemnitz, Mathias Hänel; MVZ Coburg, Coburg, Christof Lamberti; Carl-Thiem-Klinikum Cottbus gGmbH, Cottbus, Martin Schmidt-Hieber; Universitätsklinikum Dresden, Dresden, Christoph Röllig; Krankenhaus Düren GmbH, Düren, Michael Flaßhove; Universitätsklinikum Erlangen-Nürnberg, Erlangen, Andreas Mackensen; Universitätsklinikum Essen, Essen, Maher Hanoun; Klinikum Frankfurt (Oder) GmbH, Frankfurt (Oder), Michael Kiehl; Klinikum der J.W. Goethe Universität, Frankfurt (Main), Hubert Serve; Klinikum Fulda, Fulda, H.-G. Höffkes; Universitätsklinikum Halle (Saale), Halle (Saale), Lutz Peter Müller; St. Barbara-Klinik Hamm, Hamm, Heinz Albert Dürk; Universitätsklinikum Heidelberg, Heidelberg, Carsten Müller-Tidow; St. Bernward Krankenhaus, Hildesheim, Ulrich Kaiser; Universitätsklinikum Jena, Jena, Andreas Hochhaus; Westpfalz-Klinikum GmbH, Kaiserslautern, Gerhard Held; Städt, Krankenhaus Kiel, Kiel, Roland Peter Repp; Universitätsklinikum Schleswig-Holstein, Kiel, Claudia Baldus; Gemeinschaftsklinikum Mittelrhein GmbH, Koblenz, Jens-Marcus Chemnitz; Universitätsklinikum Leipzig, Leipzig, Uwe Platzbecker; Klinikum Mannheim GmbH, Mannheim, Stefan Klein; Universitätsklinikum Marburg GmbH, Marburg, Andreas Neubauer; Universitätsklinikum Münster, Münster, Wolfgang E. Berdel; Klinikum Nürnberg Nord, Nürnberg, Martin Wilhelm; Elblandklinikum Riesa, Riesa, Jörg Schubert; Agaplesion Diakonieklinikum Rotenburg GmbH, Rotenburg (Wümme), Achim Meinhardt; Diakonie-Krankenhaus, Schwäbisch-Hall, Thomas Geer; Klinikum Sindelfingen-Böblingen, Sindelfingen, Markus Ritter; Robert-Bosch-Krankenhaus, Stuttgart, Walter Erich Aulitzky; HSK Wiesbaden, Wiesbaden, Natalia Heinz; Rems-Murr-Klinikum Winnenden, Winnenden, Markus Schaich; Universitätsklinikum Würzburg, Würzburg, Hermann Einsele.

