

Key Points
Patients with VEXAS syndrome have a propensity toward developing cytopenia, MDS, multiple myeloma, and venous thromboembolism.
BM from patients with VEXAS shows characteristic vacuolization of myeloid and erythroid precursors.

Abstract
Somatic mutations in UBA1 involving hematopoietic stem and myeloid cells have been reported in patients with the newly defined VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. Here, we report clinical hematologic manifestations and unique bone marrow (BM) features in 16 patients with VEXAS. All patients were male and had a history of severe autoinflammatory and rheumatologic manifestations and a somatic UBA1 mutation (p.Met41). Ten patients had hematologic disorders: myelodysplastic syndrome (MDS; 6 of 16), multiple myeloma (2 of 16), monoclonal gammopathy of undetermined significance (2 of 16), and monoclonal B-cell lymphocytosis (2 of 16), and a few of those patients had 2 co-existing clonal processes. Although macrocytic anemia (100%) and lymphopenia (80%) were prevalent in all patients with VEXAS, thrombocytopenia and neutropenia were more common in patients with progression to MDS. All BMs in VEXAS patients had prominent cytoplasmic vacuoles in myeloid and erythroid precursors. In addition, most BMs were hypercellular with myeloid hyperplasia, erythroid hypoplasia, and varying degrees of dysplasia. All patients diagnosed with MDS were lower risk (low blast count, very good to intermediate cytogenetics) according to standard prognostic scoring with no known progression to leukemia. In addition, 10 of 16 patients had thrombotic events, including venous thromboembolism and arterial stroke. Although VEXAS presents symptomatically as a rheumatologic disease, morbidity and mortality are associated with progression to hematologic disease. Given the increased risk of developing MDS and multiple myeloma, surveillance for disease progression is important.
Introduction
VEXAS syndrome is a new disease with severe rheumatologic and hematologic manifestations and a genetic pathophysiology of somatic mutations in a single gene of critical importance in ubiquitylation. VEXAS stands for “vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic” syndrome.1 Patients present during adulthood with overlapping and variable autoinflammatory clinical manifestations, including recurrent fever, alveolitis, ear and nose chondritis, and skin lesions that may also meet diagnostic criteria for relapsing polychondritis, Sweet syndrome, polyarteritis nodosa, or giant cell arteritis. Symptoms are debilitating and refractory to treatment other than with high-dose glucocorticoids. Hematologic manifestations are present in most patients with VEXAS syndrome and include macrocytic anemia, venous and arterial thrombosis, and a propensity toward developing myelodysplasia, and plasma cell dyscrasias. The presence of abundant and coarse cytoplasmic vacuoles in myeloid and erythroid precursor cells in the bone marrow (BM) is a hallmark finding for this novel syndrome.1
Systemic autoinflammatory and autoimmune disorders have historically been associated with myeloid and lymphoid neoplasia, plasma cell dyscrasias, and thromboses. Autoinflammatory or autoimmune disorders that co-exist with myelodysplastic syndrome (MDS) were first described in the 1990s, with skin vasculitis, fevers, and arthritis being the most common nonhematologic manifestations.2,3 Since then, many retrospective studies have shown that 10% to 25% of patients with MDS have a wide clinical spectrum of autoimmunity and autoinflammation symptoms,2-10 with systemic vasculitis identified as a poor prognostic factor.2,4 Chronic inflammation, antigenic stimulation, and immune dysregulation may explain this link.11-17 The increased incidence of thrombosis in immune and inflammatory diseases is multifactorial18-24 and likely relates to activation of the coagulation cascade by cytokines and acute phase reactants as well as the presence of antiphospholipid antibodies.19,25-27
The development of genomic sequencing technologies has led to increased identification of new genes implicated in inflammatory syndromes28,29 and other nonmalignant disorders. Paroxysmal nocturnal hemoglobinuria (PNH) is the prototype of a classic hematologic syndrome later determined to be caused by a somatic mutation in the phosphatidylinositol glycan anchor biosynthesis class A (PIGA) gene. Now VEXAS, a newly discovered syndrome driven by somatic mutations in UBA1, has been identified as a link between autoinflammatory and benign and malignant hematologic disease.
Since the initial publication describing VEXAS syndrome, many centers have confirmed the inflammatory and hematologic clinical and morphological phenotype in their cohorts.30-32 Two recent studies have identified novel UBA1 variants with similar clinical manifestations,30,31 and other reports described spondyloarthritis and Kikuchi-Fujimoto disease as manifestations of VEXAS,33,34 both of which highlight the evolving understanding of this disease.33,34 In this article, we provide a comprehensive description of clinical hematologic manifestations as well as pertinent hematopathology findings in 16 patients with VEXAS syndrome.
Methods
Selection of patients
All patients were seen at the National Institutes of Health (NIH) Clinical Center, gave written informed consent, and were enrolled in protocols (NCT02257866, NCT00001373) approved by the Institutional Review Board at the NIH in accordance with the Declaration of Helsinki. Patients with confirmed UBA1 somatic mutations and clinical features of VEXAS who were evaluated at the NIH at least once and had a BM evaluation at any timepoint were included. Clinical data were obtained from NIH electronic medical records and outside records. BM specimens were reviewed by at least 2 hematopathologists. UBA1 mutation was confirmed by using whole-exome sequencing and Clinical Laboratory Improvement Amendments (CLIA)–certified Sanger sequencing as previously described.1 Standard clinical cytogenetic analyses were obtained on BM aspirates. CLIA-certified targeted next-generation sequencing for recurrently mutated genes in myeloid malignancy was performed for 9 patients. Universal Patient Number 1 (UPN-1) through UPN-15 were reported in Beck et al.1 Further details are provided in supplemental Methods.
Peripheral blood smear, BM biopsy morphology, immunohistochemistry, in situ hybridization, and flow cytometry analysis
The BM biopsies were fixed in B-plus fixative, decalcified in Rapid-Cal-Immuno (BBC Biochemical), and paraffin embedded using a Tissue-Tek processor (Sakura Finetek). Sections of 4 μm each were cut and stained using hematoxylin-and-eosin and immunohistochemical stains (CD34, CD61, myeloperoxidase, CD71, CD20, CD3, CD138, cyclin D1, CD56) as previously described35 using the Ventana BenchMark Ultra system (Ventana Medical Systems). In situ hybridization (ISH) for kappa and lambda light chains was performed to assess clonal plasma cells using the Roche BenchMark Ultra system according to manufacturer’s recommendations. Images of BM and peripheral blood (PB) were taken on a BX41 Olympus microscope with an Olympus DP74 camera using Olympus CellSens Entry 1.18 acquisition software. CellaVision DM96 Digital microscopy was used to capture PB cells where indicated. For flow cytometry, BM aspirates were stained using a panel of antibodies as previously reported35 (supplemental Methods).
Definitions
Transfusion dependence was defined as persistent red blood cell (RBC) transfusion requirement for ≥12 weeks. Thrombocytopenia was defined as mild (<100 × 103 platelets per µL), moderate (<50 × 103 platelets per µL), or severe (<20 × 103 platelets per µL). Absolute neutrophil count (ANC) <1.8 × 103 cells per µL was considered neutropenia and <0.5 × 103 cells per µL was considered severe neutropenia. Monocytopenia and lymphocytopenia were defined as lower than institutional ranges: <0.24 × 103 cells per µL (range, 0.24 × 103 to 0.86 × 103 cells per µL ) and <1.18 × 103 cells per µL (range, 1.18 × 103 to 3.74 × 103 cells per µL), respectively. World Health Organization (WHO) 2016 criteria were used to diagnose and classify MDS, and International Working Group criteria were used to diagnose and classify plasma cell neoplasm.36
Results
All 16 patients were males with a median age of 57 years (range, 45-77 years) at disease onset. Novel somatic variants at codon methionine 41 (p.Met41) of the X-linked gene UBA1 were detected in all patients, including p.Met41Thr (c.122 T>C) (50%), p.Met41Val (c.121 A>G) (31%), and p.Met41Leu (c.121 A>C) (19%). Multiorgan systemic inflammatory symptoms were present in all patients, with the most common being fatigue (100%), recurrent fevers (88%), pulmonary infiltrates (87%), skin lesions (88%), ear chondritis (73%), nose chondritis (47%), and vasculitis (67%) (supplemental Table 1). Family history was negative in patients for whom this was assessed in detail for family members with similar features.
Hematologic disorders associated with somatic UBA1 mutation
Macrocytic anemia was seen in all patients (16 of 16), and thrombocytopenia was observed in 50% (8 of 16). All patients had recorded normal vitamin B12 and folate levels, and 9 patients had copper levels tested and were normal. Although neutropenia was less common (2 [13%] of 16), absolute lymphopenia was noted in 80% of patients, and monocytopenia was noted in 50% of patients. Myeloid malignancy was diagnosed in 6 (38%) of 16 patients with VEXAS, 4 with MDS-MLD and 2 with MDS-SLD (Table 1). Median time from symptom onset to MDS was 5.0 years (range, 0.7-10.2 years). At the time of MDS diagnosis, 5/6 (83%) patients were dependent on RBC transfusions, and 1 had worsening anemia despite erythropoietin-stimulating agents (ESAs). The median platelet count was 43 × 103/µL (range, 36 × 103 to 112 × 103/µL), and median ANC was 1.51 × 103 cells per µL (range, 1.08 × 103 to 8.72 × 103cells per µL). In contrast, only 4 (40%) of 10 of those who did not meet WHO criteria for MDS were dependent on RBC transfusions. Compared with patients who met WHO criteria for MDS, those who did not had a significantly higher median platelet count of 131 × 103/µL (range, 60 × 103 to 486 × 103/µL) vs 43 × 103/µL (range, 36 × 103 to 112 × 103/µL) (P = .003; Figure 1A). Two of the 10 patients without MDS were diagnosed with multiple myeloma (MM). The median time from onset of inflammatory symptoms to last BM evaluation in the remaining 8 patients with macrocytic anemia but without an MDS diagnosis was 2.7 years (range, 1.8-9.2 years).
Details of patient and disease characteristics in VEXAS syndrome
| UPN | Age at disease onset (y) | UBA1 variant p.Met41 | UBA1 mutation VAF (%) | Hematologic diagnosis* | Thrombosis | Macrocytic anemia | RBC transfusion dependence | Hemoglobin (g/dL) | Platelets × 103/µL | ANC × 103 /µL | ALC × 103/ µL | Dysplasia on BM aspirate (%) | Marrow cellularity (%) | Precursors with cytoplasmic vacuoles† | M:E ratio | Outcome | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Malignant | Premalignant | VTE | >1 VTE | Arterial | Megakaryocyte | Myeloid | Erythroid | ||||||||||||||
| 3 | 71 | c.122 T<C; p.Met41Thr | 62.5 | MDS-MLD | — | Y | Y | Y | 7.9 | 36 | 1.08 | 0.44 | >10 | <10 | >10 | 60 | Y | 10:1 | DOD | ||
| 6 | 56 | c.122 T<C; p.Met41Val | 77.6 | MDS-MLD | — | Y | Y | Y | Y | 10.2 | 43 | 1.37 | 0.33 | >10 | >10 | <10 | 60 | Y | 5:1 | DOD | |
| 10 | 64 | c.121 T<C; p.Met41Thr | 73.1 | MDS-MLD | MGUS (IgG kappa) | Y | N | 9.9 | 46 | 1.24 | 0.85 | >10 | >10 | >10 | 95 | Y | 7:1 | Alive | |||
| 11 | 70 | c.121 A<C; p.Met41Leu | 32.5 | MDS-SLD | — | Y | Y | Y | Y | 9.9 | 42 | 1.65 | 2.84 | 10 | <10 | <10 | 90 | Y | 7:1 | Alive | |
| 14 | 56 | c.122 T<C; p.Met41Thr | 89.3 | MDS-SLD | — | Y | Y | 8.1 | 36 | 3.84 | 0.57 | 10 | <10 | <10 | 90 | Y | 8:1 | Alive | |||
| 15 | 77 | c.121 A<C; pMet41Val | 82.6 | MDS-MLD | — | Y | Y | Y | Y | 7.9 | 112 | 8.72 | 1.13 | >10 | >10 | <10 | 60 | Y | 7:1 | DOD | |
| 12 | 64 | c.121 A<C; p.Met41Leu | 76.2 | MM (IgG kappa) | MBL | Y | Y | 11.9 | 148 | 3.8 | 0.36 | <10 | <10 | <10 | 50 | Y | 4:1 | Alive | |||
| 16 | 69 | c.121 A<C; p.Met41Leu | 86.2‡ | MM (biclonal)§ | — | Y | N | 9.8 | 72 | 1.93 | 0.32 | <10 | <10 | <10 | 100 | Y | 5:1 | Alive | |||
| 1 | 45 | c.122 T<C; p.Met41Thr | 54.9 | — | — | Y | Y | Y | N | 11.1 | 79 | 2.64 | 0.21 | <10 | <10 | <10 | 60 | Y | 3:1 | DOD | |
| 2 | 56 | c.121 A<G; p.Met41Val | 52.8 | — | MGUS (IgG kappa) | Y | Y | Y | N | 13.9 | 428 | 12.1 | 0.14 | <10 | <10 | <10 | 75 | Y | 4:1 | DOD | |
| 4 | 55 | c.121 A<G; p.Met41Val | 80.5 | — | — | Y | Y | Y | Y | 10.2 | 200 | 6.2 | 0.22 | <10 | <10 | <10 | 100 | Y | 10:1 | DOD | |
| 5 | 56 | c.122 T<C; p.Met41Thr | 97.2 | — | — | Y | Y | 8.4 | 60 | 9.08 | 0.61 | <10 | <10 | <10 | 80 | Y | 7:1 | DOD | |||
| 7 | 64 | c.122 T<C; p.Met41Thr | 85.9 | — | — | Y | Y | N | 11.4 | 159 | 3.77 | 0.24 | <10 | <10 | <10 | 70 | Y | 4:1 | Alive | ||
| 8 | 53 | c.122 T<C; p.Met41Thr | 69.9 | — | — | Y | Y | Y | Y | 8.5 | 115 | 4.08 | 0.29 | <10 | <10 | <10 | 65 | Y | 4:1 | Alive | |
| 9 | 58 | c.121 A<G; p.Met41Val | 16.2 | — | — | Y | N | 12.4 | 224 | 2.45 | 0.10 | <10 | <10 | <10 | 25 | Y | 2:1 | DOD | |||
| 13 | 56 | c.122 T<C; p.Met41Thr | 68.1 | — | MBL | Y | Y | N | 8.9 | 96 | 6.22 | 0.58 | <10 | <10 | <10 | 75 | Y | 4:1 | Alive | ||
| UPN | Age at disease onset (y) | UBA1 variant p.Met41 | UBA1 mutation VAF (%) | Hematologic diagnosis* | Thrombosis | Macrocytic anemia | RBC transfusion dependence | Hemoglobin (g/dL) | Platelets × 103/µL | ANC × 103 /µL | ALC × 103/ µL | Dysplasia on BM aspirate (%) | Marrow cellularity (%) | Precursors with cytoplasmic vacuoles† | M:E ratio | Outcome | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Malignant | Premalignant | VTE | >1 VTE | Arterial | Megakaryocyte | Myeloid | Erythroid | ||||||||||||||
| 3 | 71 | c.122 T<C; p.Met41Thr | 62.5 | MDS-MLD | — | Y | Y | Y | 7.9 | 36 | 1.08 | 0.44 | >10 | <10 | >10 | 60 | Y | 10:1 | DOD | ||
| 6 | 56 | c.122 T<C; p.Met41Val | 77.6 | MDS-MLD | — | Y | Y | Y | Y | 10.2 | 43 | 1.37 | 0.33 | >10 | >10 | <10 | 60 | Y | 5:1 | DOD | |
| 10 | 64 | c.121 T<C; p.Met41Thr | 73.1 | MDS-MLD | MGUS (IgG kappa) | Y | N | 9.9 | 46 | 1.24 | 0.85 | >10 | >10 | >10 | 95 | Y | 7:1 | Alive | |||
| 11 | 70 | c.121 A<C; p.Met41Leu | 32.5 | MDS-SLD | — | Y | Y | Y | Y | 9.9 | 42 | 1.65 | 2.84 | 10 | <10 | <10 | 90 | Y | 7:1 | Alive | |
| 14 | 56 | c.122 T<C; p.Met41Thr | 89.3 | MDS-SLD | — | Y | Y | 8.1 | 36 | 3.84 | 0.57 | 10 | <10 | <10 | 90 | Y | 8:1 | Alive | |||
| 15 | 77 | c.121 A<C; pMet41Val | 82.6 | MDS-MLD | — | Y | Y | Y | Y | 7.9 | 112 | 8.72 | 1.13 | >10 | >10 | <10 | 60 | Y | 7:1 | DOD | |
| 12 | 64 | c.121 A<C; p.Met41Leu | 76.2 | MM (IgG kappa) | MBL | Y | Y | 11.9 | 148 | 3.8 | 0.36 | <10 | <10 | <10 | 50 | Y | 4:1 | Alive | |||
| 16 | 69 | c.121 A<C; p.Met41Leu | 86.2‡ | MM (biclonal)§ | — | Y | N | 9.8 | 72 | 1.93 | 0.32 | <10 | <10 | <10 | 100 | Y | 5:1 | Alive | |||
| 1 | 45 | c.122 T<C; p.Met41Thr | 54.9 | — | — | Y | Y | Y | N | 11.1 | 79 | 2.64 | 0.21 | <10 | <10 | <10 | 60 | Y | 3:1 | DOD | |
| 2 | 56 | c.121 A<G; p.Met41Val | 52.8 | — | MGUS (IgG kappa) | Y | Y | Y | N | 13.9 | 428 | 12.1 | 0.14 | <10 | <10 | <10 | 75 | Y | 4:1 | DOD | |
| 4 | 55 | c.121 A<G; p.Met41Val | 80.5 | — | — | Y | Y | Y | Y | 10.2 | 200 | 6.2 | 0.22 | <10 | <10 | <10 | 100 | Y | 10:1 | DOD | |
| 5 | 56 | c.122 T<C; p.Met41Thr | 97.2 | — | — | Y | Y | 8.4 | 60 | 9.08 | 0.61 | <10 | <10 | <10 | 80 | Y | 7:1 | DOD | |||
| 7 | 64 | c.122 T<C; p.Met41Thr | 85.9 | — | — | Y | Y | N | 11.4 | 159 | 3.77 | 0.24 | <10 | <10 | <10 | 70 | Y | 4:1 | Alive | ||
| 8 | 53 | c.122 T<C; p.Met41Thr | 69.9 | — | — | Y | Y | Y | Y | 8.5 | 115 | 4.08 | 0.29 | <10 | <10 | <10 | 65 | Y | 4:1 | Alive | |
| 9 | 58 | c.121 A<G; p.Met41Val | 16.2 | — | — | Y | N | 12.4 | 224 | 2.45 | 0.10 | <10 | <10 | <10 | 25 | Y | 2:1 | DOD | |||
| 13 | 56 | c.122 T<C; p.Met41Thr | 68.1 | — | MBL | Y | Y | N | 8.9 | 96 | 6.22 | 0.58 | <10 | <10 | <10 | 75 | Y | 4:1 | Alive | ||
ALC, absolute lymphocyte count; DOD, died of disease; MBL, monoclonal B-cell lymphocytosis; N, no; UPN, unique patient number; VTE, venous thromboembolism (confirmed by imaging); Y, yes.
Diagnosis based on 2016 WHO classification for myeloid neoplasms and International Working Group classification for plasma cell dyscrasia.
Cytoplasmic vacuoles were observed in erythroid and myeloid precursor cells.
VAF pretransplant, post-autologous transplant, 12.2%.
IgA lambda and IgG kappa.
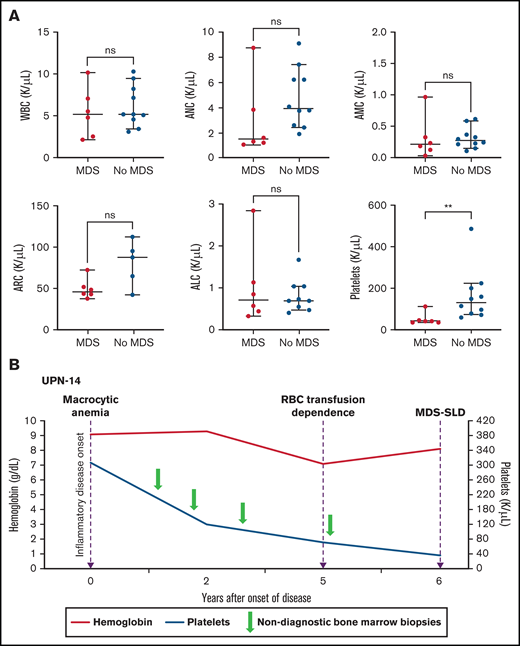
Thrombocytopenia and the risk for MDS in patients with VEXAS. (A) PB counts (from the same timepoint) are compared between patients with MDS diagnosis on BM biopsy and those without MDS diagnosis (at either the last BM biopsy performed or the biopsy before treatment for patients with MM). Median platelet counts at the time of MDS diagnosis were significantly lower (P value 0.0030) compared with those of patients whose BM was not diagnostic of MDS (this included 2 patients with MM). The 2 groups were compared using nonparametric Student t test. The error bars represent the range and middle horizontal bars mark the median. (B) Serial blood counts and BM evaluations for UPN-14 are used as an example to show disease progression. Macrocytic anemia was seen at the onset of inflammatory disease, which progressed over the next 5 years to RBC transfusion dependence. After 5 years, there was steady decline in platelet counts to <50 × 103/µL 1 year later, when the BM biopsy revealed diagnosis of MDS-SLD. Previous BM evaluations did not reveal a diagnosis. ALC, absolute lymphocyte count; AMC, absolute monocyte count; ARC, absolute reticulocyte count.
Thrombocytopenia and the risk for MDS in patients with VEXAS. (A) PB counts (from the same timepoint) are compared between patients with MDS diagnosis on BM biopsy and those without MDS diagnosis (at either the last BM biopsy performed or the biopsy before treatment for patients with MM). Median platelet counts at the time of MDS diagnosis were significantly lower (P value 0.0030) compared with those of patients whose BM was not diagnostic of MDS (this included 2 patients with MM). The 2 groups were compared using nonparametric Student t test. The error bars represent the range and middle horizontal bars mark the median. (B) Serial blood counts and BM evaluations for UPN-14 are used as an example to show disease progression. Macrocytic anemia was seen at the onset of inflammatory disease, which progressed over the next 5 years to RBC transfusion dependence. After 5 years, there was steady decline in platelet counts to <50 × 103/µL 1 year later, when the BM biopsy revealed diagnosis of MDS-SLD. Previous BM evaluations did not reveal a diagnosis. ALC, absolute lymphocyte count; AMC, absolute monocyte count; ARC, absolute reticulocyte count.
Characteristic PB and BM findings in patients with a UBA1 mutation
On review of peripheral smears, all patients had evidence of RBC macrocytosis ranging from mild to moderate. Circulating immature granulocytic precursors were found in 11 of 16 patients (supplemental Figure 1A-B; supplemental Table 2). Ten patients had 1 or more of the following: cytoplasmic vacuoles and hypogranular or hyposegmented neutrophils, some with pseudo-Pelger-Huet–like morphology (supplemental Figure 1C-H). Vacuolated monocytes were observed in 9 patients (supplemental Figure 1I-J).
BM aspirate smears showed marked cytoplasmic vacuolization in hematopoietic precursors, including blasts and erythroid and myeloid precursors, in all evaluated patients (Figure 2D-J; supplemental Table 3). The average percentage of myeloid and erythroid cells with vacuoles was ∼15% with an average of 5 to 7 vacuoles per cell. Of note, vacuoles were predominantly found in early precursors (blasts, promyelocytes, and pronormoblasts). Vacuoles were also identified in eosinophils, monocytes, plasma cells, and megakaryocytes to a lesser degree (Figure 2K-M). Lymphocytes were generally devoid of vacuoles.
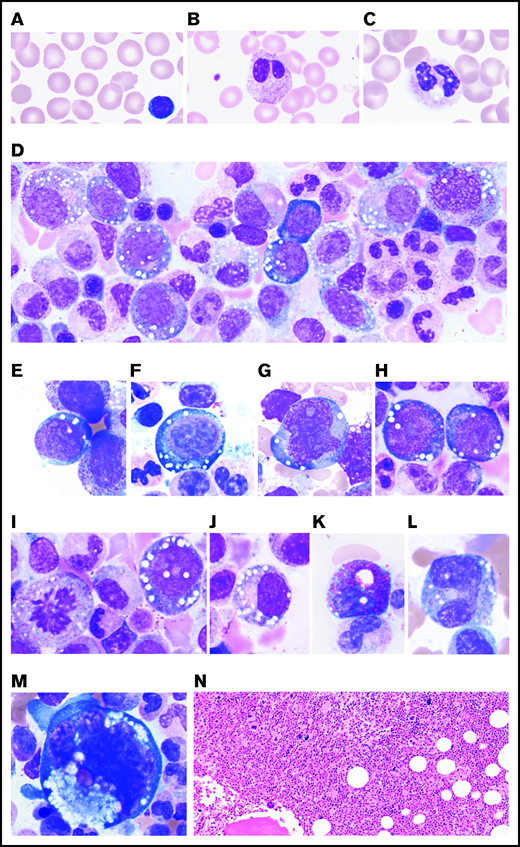
PB and BM features of VEXAS. All patients studied had (A) macrocytic anemia demonstrated by RBCs greater in size than the nuclei of small lymphocytes. (B) Circulating hyposegmented pelgeroid neutrophils and (C) vacuolated neutrophils are common. Bone BM aspirates show striking vacuolization of (D) myeloid and erythroid precursors including (E) blasts, (F) erythroid pronormoblasts, (G) monoblasts, (H-I) promyelocytes, (J) myelocytes, (K) eosinophilic myelocytes, (L) promonocytes, and less commonly (M) megakaryocytes. (N) Core biopsies typically demonstrate hypercellular BM with myeloid hyperplasia (hematoxylin and eosin (H&E) stain; original magnification ×200). (A-M) Wright-Giemsa stained smears; original magnification ×1000.
PB and BM features of VEXAS. All patients studied had (A) macrocytic anemia demonstrated by RBCs greater in size than the nuclei of small lymphocytes. (B) Circulating hyposegmented pelgeroid neutrophils and (C) vacuolated neutrophils are common. Bone BM aspirates show striking vacuolization of (D) myeloid and erythroid precursors including (E) blasts, (F) erythroid pronormoblasts, (G) monoblasts, (H-I) promyelocytes, (J) myelocytes, (K) eosinophilic myelocytes, (L) promonocytes, and less commonly (M) megakaryocytes. (N) Core biopsies typically demonstrate hypercellular BM with myeloid hyperplasia (hematoxylin and eosin (H&E) stain; original magnification ×200). (A-M) Wright-Giemsa stained smears; original magnification ×1000.
Half the patients had BM evaluation at multiple time points, and the other half had BM evaluation just once. Findings from the most recent or the most recent pretreatment BM evaluation are summarized in Table 1. The BM was hypercellular in 14 (87.5%) of 16 patients (Figure 2N) and ranged in cellularity from 25% to 100% (interquartile range [IQR], 60%-90%) with a median of 70% cellularity. More than half the patients (9 of 16) showed myeloid hyperplasia with myeloid:erythroid (M:E) ratios of 7:1 or greater, more frequently observed in patients (5 of 6) with MDS. The remaining patients showed mild myeloid hyperplasia or had normal M:E ratios (Table 1). In the 8 patients with multiple BM specimens, the cellularity increased over time in 4 patients, with a median increase of 20% cellularity (IQR, 16%-35%) over a median time of 3.5 years (IQR, 1.9-7.0 years). No patients demonstrated loss of BM cellularity without treatment. BM blast count was <5% in all BM specimens. Megakaryocytes were increased in half the BMs and decreased in 2 of 16 BMs.
Flow cytometric data were available for BM aspirates from 11 patients (supplemental Table 4; supplemental Figure 3). The results revealed several unusual features: absent B-cell precursors (<1% of lymphocytes) in 10 (91%) of 11, inverted CD4:CD8 T-cell ratio in 8 (73%) of 11, and an aberrant CD56 expression on >10% of monocytes in 6 of 10 (supplemental Figure 3). Low numbers of natural killer cells were observed in 2 patients. Ten of 11 patients had relatively increased numbers of CD57+ T cells representing >10% of lymphocytes, and 5 of 11 had >20%, which is consistent with increased large granular lymphocytes. Clonal B-cell populations indicative of monoclonal B-cell lymphocytosis were observed in 2 (13%) of 16 patients, both with immunophenotypic features characteristic of chronic lymphocytic leukemia (CD5+ and CD23+). In PB, CD19 B cells were low in 8 of 12 patients, whereas natural killer cells were decreased in 10 of 12 patients (supplemental Table 5).
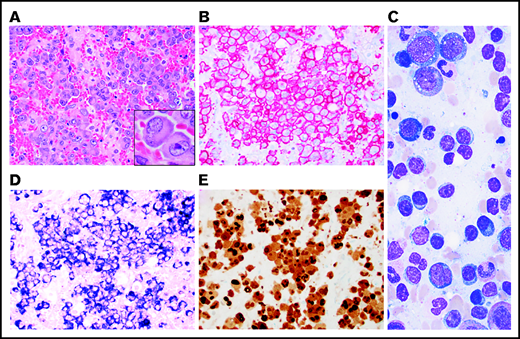
Some degree of atypia or dyspoiesis in megakaryocytes and myeloid and erythroid precursors (Table 1) were present in nearly all BM aspirates, but dysplasia in >10% of the cells in a lineage was seen only in those diagnosed with MDS. All patients with an MDS diagnosis had hypercellular BM with an increased M:E ratio, myeloid hyperplasia, and erythroid hypoplasia. All had evidence of megakaryocytic dysplasia characterized by hypolobated or mononuclear megakaryocytes, megakaryocytes with separated nuclear lobes, and/or micromegakaryocytes highlighted by CD61 IHC (Figure 3A-E). Of the 4 patients who met criteria for MDS-MLD, 2 had evidence of dyserythropoiesis demonstrated by binucleation or multinucleation, nuclear budding, and/or marked megaloblastic changes, and 3 had evidence of dysmyelopoisis with hypogranular and/or hyposegmented precursors (Figure 3E-J).

MDS in patients with VEXAS. MDS in patients with VEXAS is associated with hypercellular BM (panel A: H&E stain; original magnification ×500) with dysplastic megakaryocytes, including micromegakaryocytes highlighted by CD61 immunohistochemistry (IHC) on the core biopsy (panel B: original magnification ×500). Dysmegakaryopoiesis can be seen on aspirate smears, including (C) micromegakaryocytes, (D) megakaryocytes with vacuoles, and (E) megakaryocytes with separated nuclear lobes. Dyserythropoiesis including (F-G) binucleation and multinucleation, megaloblastic changes, and nuclear budding are common. Dysmyelopoiesis can be striking with abnormal maturation including (G) binucleation and maturation asynchrony, (H) binucleated eosinophilic myelocytes, (I) hypogranular forms, and (J) myeloid cells with abnormal morphology and vacuoles. All aspirate images show Wright-Giemsa–stained smears; original magnification ×1000.
MDS in patients with VEXAS. MDS in patients with VEXAS is associated with hypercellular BM (panel A: H&E stain; original magnification ×500) with dysplastic megakaryocytes, including micromegakaryocytes highlighted by CD61 immunohistochemistry (IHC) on the core biopsy (panel B: original magnification ×500). Dysmegakaryopoiesis can be seen on aspirate smears, including (C) micromegakaryocytes, (D) megakaryocytes with vacuoles, and (E) megakaryocytes with separated nuclear lobes. Dyserythropoiesis including (F-G) binucleation and multinucleation, megaloblastic changes, and nuclear budding are common. Dysmyelopoiesis can be striking with abnormal maturation including (G) binucleation and maturation asynchrony, (H) binucleated eosinophilic myelocytes, (I) hypogranular forms, and (J) myeloid cells with abnormal morphology and vacuoles. All aspirate images show Wright-Giemsa–stained smears; original magnification ×1000.
Half of MDS patients had abnormal cytogenetics: UPN-6 had successive acquisition of abnormal clones with the last BM evaluation showing del5q and del13q; UPN-15 had del20q, and UPN-3 had t(3;12). Four of the 6 patients with MDS diagnosis had next-generation sequencing for 177 recurrently mutated genes in hematologic malignancies, and 3 were found to have somatic variants: 2 with DNMT3A (variant allele frequency [VAF], 43% and 36%), and 1 with GNA11 (VAF, 3.3%) and CSF1R (VAF, 3.1%). In addition, EZH2 (VAF, 21.13%) was found in 1 patient (UPN-1) without an MDS diagnosis (Table 2).
Plasma cell neoplasia was diagnosed in 4 patients (25%) with VEXAS, including 2 with MM (Figure 4) and 2 with monoclonal gammopathy of undetermined significance (MGUS) (supplemental Figure 3; supplemental Table 6). UPN-12 had 2 pretreatment BM evaluations performed to investigate macrocytic anemia; both showed ∼10% to 20% clonal plasma cells. UPN-16 had a BM evaluation for anemia and thrombocytopenia that showed MM with 70% clonal plasma cells, t(11;14) by fluorescence in situ hybridization and amyloid deposition on Congo Red staining confirmed by liquid chromatography-tandem mass spectrometry. Three patients had IgG kappa paraprotein, and 1 MM patient (UPN-16) had biclonal IgA lambda and IgG kappa paraproteins. Interestingly, UPN-10 had both MGUS with 8% monoclonal plasma cells in BM and MDS-MLD (supplemental Figure 3). UPN-12 had MM and an abnormal monoclonal B-cell lymphocyte population.
MM in patients with VEXAS. Extensive BM infiltration by markedly atypical plasma cells on core biopsy (panel A: H&E stain; original magnification ×500, inset ×1000 ), highlighted by CD138 IHC (panel B: original magnification ×500). Marrow aspirate showed myeloid precursors with vacuoles and increased scattered plasma cells (panel C: Wright-Giemsa stain; original magnification ×1000). The plasma cells expressed lambda light chains by in situ hybridization on core biopsy (panel D; original magnification ×500) and aberrant expression of cyclin D1 by IHC (panel E: original magnification ×500).
MM in patients with VEXAS. Extensive BM infiltration by markedly atypical plasma cells on core biopsy (panel A: H&E stain; original magnification ×500, inset ×1000 ), highlighted by CD138 IHC (panel B: original magnification ×500). Marrow aspirate showed myeloid precursors with vacuoles and increased scattered plasma cells (panel C: Wright-Giemsa stain; original magnification ×1000). The plasma cells expressed lambda light chains by in situ hybridization on core biopsy (panel D; original magnification ×500) and aberrant expression of cyclin D1 by IHC (panel E: original magnification ×500).
Progression of BM disease
BM slides from 8 of 16 patients were available for review at multiple timepoints. Cytoplasmic vacuolization of myeloid and erythroid precursors was present in all BM aspirates, regardless of disease timing. Disease progression in the BM was documented in 4 patients (UPN-3, UPN-4, UPN-6, UPN-10); all had multiple inflammatory manifestations at disease onset (Table 2). All 4 had initial BM evaluation for macrocytic anemia. BMs were either normocellular (n = 2) or hypercellular (n = 2) with myeloid hyperplasia but without overt dysplasia. Repeat BM evaluations were performed at varying times from the initial BM evaluation because of worsening cytopenia; 3 patients progressed to transfusion-dependent anemia, and all developed severe thrombocytopenia (platelets <50 × 103/µL). BMs were hypercellular with myeloid hyperplasia and erythroid hypoplasia in all patients. Dyspoiesis was more pronounced than the observed level in previous BMs in all 4 patients; 3 met WHO criteria for MDS-MLD. Abnormal cytogenetics were identified in 2 patients (UPN-3 and UPN-6; Table 1). Although not all BM slides were available for review, UPN-14 displayed a similar pattern of progressive cytopenia with eventual diagnosis of MDS (Figure 1B).
Progression of disease in patients with available sequential BM evaluations
| UPN | Inflammatory manifestations | Years from disease onset | WBC × 103/µL | Hb g/dL | MCV fL | Platelets × 103/µL | Marrow evaluation | M:E ratio | Cytogenetics | Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | Fever, joint effusion, synovitis, spondyloarthropathy, hearing loss | 1 | 2.89 | 10.5 | 9.2 | 259 | Normocellular, relative myeloid hyperplasia | 4:1 | Normal | — |
| 4 | 7.03 | 7.9 | 109.9 | 36 | Hypercellular, megakaryocytic and erythroid lineage dysplasia, erythroid hypoplasia, myeloid hyperplasia | 10:1 | t(3:12)(q21;q13) [17/20] | MDS-MLD | ||
| 4 | Fever, rash, medium vessel vasculitis | 3 | 7.13 | 10.2 | 103 | 200 | Normocellular, myeloid hyperplasia, erythroid hypoplasia | 7:1 | Normal | — |
| 6 | 16 | 8.9 | 109.4 | 38 | Hypercellular (100%), myeloid hyperplasia, erythroid hypoplasia, <10% dyspoiesis of myeloid and erythroid precursors | 10:1 | Normal | — | ||
| 6 | Fever, rash, vasculitis, Sweet’s syndrome | 3 | 4.08 | 8.8 | 109.4 | 213 | Normocellular, trilineage hematopoiesis | 4:1 | Normal | — |
| 10 | 2.1 | 10.2 | 102 | 43 | Hypercellular with megakaryocytic and myeloid dysplasia, erythroid hypoplasia | 10:1 | (46,XY,del(5) (q22q33), del(13)(q12q14) [16]/46,XY[4] | MDS-MLD | ||
| 10 | Sweet’s syndrome, ear chondritis, rash | 3 | 5.02 | 9.6 | 111.8 | 110 | Hypercellular with mild dyspoietic features, 8% clonal plasma cells | Normal | — | |
| 4.5 | 2.51 | 9.9 | 116.9 | 46 | Hypercellular, dysplasia in all 3 lineages, 8% clonal plasma cells | Normal | MDS-MLD | |||
| 1 | Fever, rash, flu-like symptoms, hearing loss, diarrhea | 9 | 3.76 | 11.1 | 110 | 108 | Normocellular | 3:1 | Normal | — |
| 10 | 3.08 | 11.1 | 109.8 | 79 | Normocellular | 3:1 | Normal | — | ||
| 8 | Fever, ear chondritis, pulmonary infiltrate, hearing loss | 2 | 3.79 | 10.4 | 129 | 166 | Mildly hypercellular | 3:1 | Normal | — |
| 3 | 5.09 | 8.5 | 125.9 | 115 | Mildly hypercellular | 4:1 | Normal | — |
| UPN | Inflammatory manifestations | Years from disease onset | WBC × 103/µL | Hb g/dL | MCV fL | Platelets × 103/µL | Marrow evaluation | M:E ratio | Cytogenetics | Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | Fever, joint effusion, synovitis, spondyloarthropathy, hearing loss | 1 | 2.89 | 10.5 | 9.2 | 259 | Normocellular, relative myeloid hyperplasia | 4:1 | Normal | — |
| 4 | 7.03 | 7.9 | 109.9 | 36 | Hypercellular, megakaryocytic and erythroid lineage dysplasia, erythroid hypoplasia, myeloid hyperplasia | 10:1 | t(3:12)(q21;q13) [17/20] | MDS-MLD | ||
| 4 | Fever, rash, medium vessel vasculitis | 3 | 7.13 | 10.2 | 103 | 200 | Normocellular, myeloid hyperplasia, erythroid hypoplasia | 7:1 | Normal | — |
| 6 | 16 | 8.9 | 109.4 | 38 | Hypercellular (100%), myeloid hyperplasia, erythroid hypoplasia, <10% dyspoiesis of myeloid and erythroid precursors | 10:1 | Normal | — | ||
| 6 | Fever, rash, vasculitis, Sweet’s syndrome | 3 | 4.08 | 8.8 | 109.4 | 213 | Normocellular, trilineage hematopoiesis | 4:1 | Normal | — |
| 10 | 2.1 | 10.2 | 102 | 43 | Hypercellular with megakaryocytic and myeloid dysplasia, erythroid hypoplasia | 10:1 | (46,XY,del(5) (q22q33), del(13)(q12q14) [16]/46,XY[4] | MDS-MLD | ||
| 10 | Sweet’s syndrome, ear chondritis, rash | 3 | 5.02 | 9.6 | 111.8 | 110 | Hypercellular with mild dyspoietic features, 8% clonal plasma cells | Normal | — | |
| 4.5 | 2.51 | 9.9 | 116.9 | 46 | Hypercellular, dysplasia in all 3 lineages, 8% clonal plasma cells | Normal | MDS-MLD | |||
| 1 | Fever, rash, flu-like symptoms, hearing loss, diarrhea | 9 | 3.76 | 11.1 | 110 | 108 | Normocellular | 3:1 | Normal | — |
| 10 | 3.08 | 11.1 | 109.8 | 79 | Normocellular | 3:1 | Normal | — | ||
| 8 | Fever, ear chondritis, pulmonary infiltrate, hearing loss | 2 | 3.79 | 10.4 | 129 | 166 | Mildly hypercellular | 3:1 | Normal | — |
| 3 | 5.09 | 8.5 | 125.9 | 115 | Mildly hypercellular | 4:1 | Normal | — |
No patients had blasts ≥5%.
Hb, hemoglobin; MCV, mean corpuscular volume; WBC, white blood cell.
UPN-1 and UPN-8 had worsening macrocytic anemia without transfusion dependence and mild thrombocytopenia; repeat BM evaluation after a year did not show significant changes. The remaining 2 patients with available sequential BM evaluations for review had diagnosis of MM made on the first BM aspiration with posttreatment follow-up BM aspirations.
Outcomes
Transformation to MDS with excess blasts or acute myeloid leukemia (AML) did not occur in any patient. Patients received ESAs, lenalidomide, and hypomethylating agents (n = 2) but did not show any improvement (Table 3). UPN-15 who had MM had no response to bortezomib and dexamethasone after 3 cycles but had a partial response to daratumumab monotherapy (with persistent clonal plasma cells of 10% in BM). He received ESAs for anemia and has remained transfusion independent. Of interest, his inflammatory symptoms initially were ameliorated after initiation of daratumumab therapy. UPN-16 achieved a very good partial response with 2 cycles of lenalidomide, bortezomib, and dexamethasone and then underwent autologous stem cell transplantation followed by bortezomib maintenance with remission and improvement in inflammatory symptoms. Unfortunately, the patient’s MM relapsed 6 months after autologous stem cell transplantation.
MDS diagnosis and treatment and patient outcome
| UPN | WHO categorization* | IPSS-R | Morphologic dysplasia lineages >10% | Cytogenetics | Somatic variant (VAF %) | Treatment | Outcome |
|---|---|---|---|---|---|---|---|
| 3 | MDS-MLD | 3.5 | Megakaryocyte, erythroid | 46,XY,t(3;12)(q21;q13)[17]/46,XY[3] | NA | NA | Died |
| 6 | MDS-MLD | 3 | Megakaryocyte, myeloid | 46,XY,del(5)(q22q33),del(13)(q12q14)[16]/46,XY[4] | DNMT3A (42.56%)† | Lenalidomide, HMA‡ | Died |
| 10 | MDS-MLD | 4 | Megakaryocyte, myeloid, erythroid | 46,XY | DNMT3A (44.37%)† | ESA | Worsening cytopenia |
| 11 | MDS-SLD | 3 | Megakaryocyte | 46,XY | GNA11 (3.3%), CSF1R (3.12%) | Supportive transfusions | Stable disease, 9 months |
| 14 | MDS-SLD | 4 | Megakaryocyte, myeloid | 46,XY | No variant | HMA × 4 cycles (NR), supportive transfusions | Stable disease, 3 months |
| 15 | MDS-MLD | 3.5 | Megakaryocyte, myeloid | 46,XY,del(20)(q11.2q13.3)[12]/46,XY[8] | NA | NA | Died |
| UPN | WHO categorization* | IPSS-R | Morphologic dysplasia lineages >10% | Cytogenetics | Somatic variant (VAF %) | Treatment | Outcome |
|---|---|---|---|---|---|---|---|
| 3 | MDS-MLD | 3.5 | Megakaryocyte, erythroid | 46,XY,t(3;12)(q21;q13)[17]/46,XY[3] | NA | NA | Died |
| 6 | MDS-MLD | 3 | Megakaryocyte, myeloid | 46,XY,del(5)(q22q33),del(13)(q12q14)[16]/46,XY[4] | DNMT3A (42.56%)† | Lenalidomide, HMA‡ | Died |
| 10 | MDS-MLD | 4 | Megakaryocyte, myeloid, erythroid | 46,XY | DNMT3A (44.37%)† | ESA | Worsening cytopenia |
| 11 | MDS-SLD | 3 | Megakaryocyte | 46,XY | GNA11 (3.3%), CSF1R (3.12%) | Supportive transfusions | Stable disease, 9 months |
| 14 | MDS-SLD | 4 | Megakaryocyte, myeloid | 46,XY | No variant | HMA × 4 cycles (NR), supportive transfusions | Stable disease, 3 months |
| 15 | MDS-MLD | 3.5 | Megakaryocyte, myeloid | 46,XY,del(20)(q11.2q13.3)[12]/46,XY[8] | NA | NA | Died |
HMA, hypomethylating agent; IPSS-R, Revised International Prognostic Scoring System; NA, not available; NR, no response.
WHO, World Health Organization 2016 classification for myeloid neoplasm
UPN-6 had DNMT3A missense variant NM_175629.2:c.2657A>G (NP_783328.1:p.Gln886Arg) and UPN-10 had DNMT3A missense variant NM_175629.2:c.2644C>T (NP_783328.1:p.Arg882Cys).
Lenalidomide was discontinued because of rash, and HMA was given for only 1 cycle because of hospitalization for inflammatory flare.
Nine (56%) of 16 patients died as a result of disease-related causes; 3 had a diagnosis of MDS, and 1 had MGUS (Table 3). Mortality differed by specific genetic variant: 5 (63%) of 8 had p.Met41Val, 4 (50%) of 8 had p.Met41Thr, and 0 (0%) of 8 had p.Met41Leu. Length of follow-up time did not differ among patients with these variants.
Thrombotic complications
Thrombotic events occurred in 10 (63%) of 16 patients; 9 had venous thromboembolism (VTE) and UPN-7 had a stroke (supplemental Table 7). Of the 9 patients with VTE, 8 had an unprovoked event and 7 had >1 VTE (range, 1-6 VTEs) with a median of 2 events per patient. Three patients had documented recurrence of VTEs while receiving therapeutic anticoagulation. Thrombotic events occurred primarily early in the disease course (range, 0-67 months); in 6 (60%) of 10 patients, the first thrombotic event occurred within the first 2 years after disease onset, and in 2 patients, it occurred at initial presentation of inflammatory disease. The only arterial thrombotic event was a stroke that occurred at 67 months after the onset of disease in the absence of documented arrhythmia or carotid stenosis.
Standard and specialized coagulation testing were performed (supplemental Tables 8 and 9) at time of NIH Clinical Center assessment. All patients were tested for lupus anticoagulant (LA). Of these, 7 (44%) of 16 patients were persistently positive for LA (6 of whom had LA tests that wet were 12 weeks apart and one patient had them 8 weeks apart) and 5 of them had a documented thrombosis. Four patients had 1 positive test for LA that was not repeated and therefore could not be called persistently positive (of these, 2 had thrombosis). Five patients tested for LA had negative results, 2 of whom had thrombosis. Fourteen patients were screened for anticardiolipin (ACA), and 5 patients were screened for β2-glycoprotein (B2GP) antibodies (both immunoglobulin G [IgG] and IgM). Tests for ACA IgG and B2GP IgG/IgM were negative in all patients, but tests for ACA IgM were weakly positive in 2 patients at 37 and 29 U/mL; both of these patients were positive for LA, and 1 had a documented thrombosis.
Factor VIII (FVIII) levels were determined in 5 patients and were high in 3 patients (214%, 249%, 279%; 2 of those patients had thrombosis and 1 did not). Patients with high FVIII levels had corresponding increased C-reactive protein levels of 11 µg/ml, 64 µg/ml, and 78 µg/ml; those with normal FVIII had levels of 6 and 50. FIX levels were assessed in 2 patients; the level was high in 1 patient (210%) who had thrombosis. FII, FV, FVII, FX, and FXI were normal in 2 tested patients. D-dimer was elevated in all 6 tested patients, although 2 had normal levels when the levels were adjusted for age. All 8 patients tested for PNH had negative results. No patients were found to have significantly abnormal von Willebrand factor activity, antithrombin III activity, or protein C or S activity; 2 patients had levels just above the normal range (UPN-12 in von Willebrand factor and UPN-1 in protein C). Thromboelastography was performed for 2 patients (1 with thrombosis), and both patients had normal values.
Discussion
VEXAS syndrome as a result of somatic mutations in UBA1 p.Met41 in hematopoietic stem and progenitor cells is a recently identified disease with inflammatory and hematologic manifestations.1 Multiorgan severe systemic autoinflammation is resistant to available treatments except high-dose glucocorticoids. Striking hematologic features include the presence of cytoplasmic vacuoles in hematopoietic precursor cells in the BMs of all patients, high rates of VTE, progressive cytopenia, or a diagnosis of MDS or plasma cell dyscrasia in 40% of initially reported patients1 that was recapitulated in subsequent follow-up studies. Herein, we have expanded on the clinical spectrum of both benign and malignant hematologic disease and characterized the pertinent BM findings in VEXAS syndrome.
Macrocytic anemia was the first sign of hematopoietic dysfunction identified in all patients. Peripheral smears in the majority of patients showed abnormal neutrophils with cytoplasmic vacuoles, hypogranularity, and hyposegmentation. Characteristic BM features included the presence of cytoplasmic vacuoles in all patients, hypercellularity, high M:E ratio, and decreased B-cell precursors. Discrete cytoplasmic vacuoles were more frequent and higher in number per cell in erythroid and myeloid precursor cells than in more mature cells. Cytoplasmic vacuoles in myeloid and erythroid precursors have been reported in MDS and in copper deficiency and autoimmune diseases.32,37 None of our patients had copper deficiency. The lymphoid lineage, which is known to have vacuoles in many malignant diseases, including Burkitt lymphoma38 and acute lymphocytic leukemia,39 was largely devoid of them in VEXAS. Somatic UBA1 mutations are restricted to the erythroid and myeloid lineage, likely explaining the location of the vacuoles’ lineage. Vacuoles were found irrespective of dysplasia, and the percentage of vacuoles was not increased in the BMs of patients who met WHO criteria for MDS. Similar BM findings have been described in 2 other novel UBA1 mutations.30,31UBA1 mutation testing should be considered when evaluating any patients with BM vacuoles along with previously established differential diagnoses, including MDS and hypocupremia.32,37
Increased T cells with cytotoxic phenotypes, including CD3+CD57+ subsets and an inverted CD4:CD8 ratio, are consistent with an acquired, activated T-cell repertoire that has been demonstrated in age-related changes, immunodeficiency states, chronic viral infections, and aplastic anemia, which support involvement of an immune mechanism in cytopenia.40,41 BM flow cytometry showed a complete loss of B-cell precursors, which is observed in patients with primary immune deficiencies and in patients with de novo MDS.42,43
Data obtained from serial laboratory tests and BM evaluations identified a clear pattern of progressive BM dysfunction. Three stages of cytopenia were apparent: (1) mild macrocytic anemia without other cytopenia, (2) progressive macrocytic anemia (not transfusion dependent) with mild thrombocytopenia, and (3) progressive transfusion-dependent macrocytic anemia and severe thrombocytopenia. Neutropenia was rare. BM biopsies that did not meet MDS diagnostic criteria were hypercellular with myeloid hyperplasia and erythroid hypoplasia (increased M:E ratio), which may represent a transition to overt malignancy or may be the result of differential intrinsic effect of UBA1 mutation on the 2 lineages. Most importantly, presence of transfusion-dependent anemia and severe thrombocytopenia (platelets <50 × 103/µL) were associated with morphologic MDS by WHO criteria. No particular UBA1 mutation correlated with MDS progression. One study limitation is the retrospective nature of some cohort patients. Although evolution to high-grade MDS or AML was not observed in our cohort, prospective and longitudinal studies are necessary to elucidate the biologic disease course in VEXAS.
On the basis of our results, somatic mutations in UBA1 have a much higher risk of progression to MDS (40%) than is typically observed in clonal hematopoiesis (4%)44 or in well-established clonal diseases like PNH (2% to 6%).45 It is not clear whether the transformation to MDS occurs as a result of primary clonal expansion from proliferation and/or survival advantage of mutated cells or is a secondary product of an altered highly inflammatory microenvironment. All patients with MDS were lower risk according to the Revised International Prognostic Scoring System (IPSS-R). To date, no progression to higher-risk MDS or AML has been observed. Only 3 recurrent myeloid cancer gene mutations in epigenetic regulators were identified in VEXAS patients: DNMT3A (n = 2) and EZH2 (n = 1). Although ESA support showed some benefit, both lenalidomide and hypomethylating agent therapy were either poorly tolerated or did not provide any benefit in the few patients who received them. No particular treatment was associated with improved outcome. Sweet syndrome is a known harbinger of MDS46; several of our patients had a diagnosis of Sweet syndrome, 2 of whom developed MDS.
Patients with VEXAS also met diagnostic criteria for plasma cell dyscrasia (2 with MM and 2 with MGUS). All had IgG kappa paraprotein (1 was biclonal with dominant IgΑ lambda), and 3 of 4 had evidence of t(11;14). Interestingly, patients who received treatment for MM also had improved inflammatory symptoms. This may be the result of concurrent glucocorticoid administration or may be the result of the inherent activity of MM-directed therapy with bortezomib or daratumumab. Although therapy-related MDS occurs in MM patients47 and MDS before or after MGUS or MM occurs,48 coexistence of de novo MDS and plasma cell dyscrasia is less widely reported.49 Patients with MGUS or MM have a higher risk than controls of developing MDS,50 suggesting an underlying link between MDS and plasma cell neoplasms, as we observed in VEXAS, in which a common molecular mechanism related to somatic mutations in UBA1 may contribute to this association.
Thrombotic complications in VEXAS seem to be common and may occur early in the disease course with later recurrence. High rates of thrombosis are reported in other autoimmune and autoinflammatory disorders.20,23 Etiologies underlying thrombosis in these disorders vary and include activation of the coagulation cascade, localized venous or arterial inflammation seen in vasculitis, neutrophil extracellular traps, and the presence of anti-phospholipid antibodies. In VEXAS syndrome, the findings of high FVIII levels, high D-dimers, and high inflammatory markers suggest activation of the coagulation cascade as one possible mechanism of hypercoagulability. The most consistent finding in VEXAS was that the majority of patients had a positive LA test that was positive on more than 1 occasion in 44%. Antiphospholipid-specific antibodies ACA and B2GP test results were almost always negative, and LA positivity may reflect the presence of other nonspecific antibodies. Localized skin and vessel wall inflammation as seen in vasculitis may also contribute to thrombosis because the majority of VEXAS patients had ear or nose chondritis, and 6 patients met diagnostic criteria for vasculitis. However, specialized coagulation testing was not available at the time of acute thrombosis for most patients and was instead performed subsequently during routine clinical evaluation. Further mechanistic studies will be required to elucidate the cause of thrombosis.
In summary, VEXAS is complex and heterogenous with many overlapping diseases, including a severe inflammatory disorder, cytopenia without a clear diagnosis, MDS, plasma cell dyscrasia, and thrombosis. Patients with VEXAS have a high morbidity because of progressive and treatment refractory inflammatory disease, as well as progressive cytopenia with or without MDS or MM. Furthermore, complications occur from prolonged glucocorticoid treatment and a requirement for chronic RBC transfusions. Multidisciplinary care led by a rheumatologist and a hematologist is required, but treatment may involve many other specialists. Because of the high mortality rate with VEXAS and lack of effective treatments, prompt diagnosis is critical; VEXAS should be suspected in patients with recalcitrant inflammation and persistent cytopenia. BM biopsy should be considered when VEXAS is suspected clinically. We present prominent clinical, laboratory, and BM features to identify these patients early and to allow early identification and consideration for investigative treatments to attempt eradication of the UBA1 clone, such as allogeneic hematopoietic stem cell transplant (HSCT).
Much remains unknown about both clinical and pathophysiological aspects of VEXAS. Prospective follow-up of patients will further illuminate the pattern of disease course, true incidence of malignant transformation, and heterogeneity in the clinical phenotype. Development of animal models will be helpful in further elucidating the biology of VEXAS and will help identify therapeutics. Although allogeneic HSCT may offer cure, this therapy is not without risk and may not be an option for all patients with VEXAS because of older age, comorbidities, or lack of suitable donors. Therefore, clinical studies evaluating both medical therapies and HSCT are crucial going forward.
Acknowledgments
The authors thank all the affected patients and their families for participating in this research study.
This work was supported by grants from the Intramural Research Program of the National Institutes of Health (NIH); NIH Clinical Center; National Human Genome Research Institute; National Heart, Lung, and Blood Institute; National Cancer Institute; and National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Authorship
Contribution: I.E.O., B.A.P., E.M.G., D.B.B., and K.R.C. designed the study, performed analyses, and wrote the manuscript; B.A.P., E.M.G., M.A.F., A.K.O., J.L., P.C.G., D.B.B., L.W., P.H. and N.S.Y. provided clinical care to patients; I.E.O., K.R.C., N.P., and A.D.-F. provided pathologic interpretations; Z.W., W.W., M.T, D.O.C., and F.G.-R. performed laboratory studies; and D.L.K., P.C.G., N.S.Y. edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Katherine R. Calvo, Hematology Section, Department of Laboratory Medicine, Clinical Center, National Institutes of Health, 10 Center Dr, Building10/Room 2C306, Bethesda, MD, 20892-1508; e-mail: calvok@cc.nih.gov.

