

Key Points
We describe the first cases of B-ALL in patients with RUNX1-FPD.
We identify a novel germline RUNX1 mutation underlying RUNX1-FPD.
Abstract
Germline RUNX1 mutations underlie a syndrome, RUNX1-familial platelet disorder (RUNX1-FPD), characterized by bleeding symptoms that result from quantitative and/or qualitative defect in platelets and a significantly increased risk for developing hematologic malignancies. Myeloid neoplasms are the most commonly diagnosed hematologic malignancies, followed by lymphoid malignancies of T-cell origin. Here, we describe the first 2 cases of B-cell acute lymphoblastic leukemia (B-ALL) in patients with confirmed germline RUNX1 mutations. While 1 of the patients had a known diagnosis of RUNX1-FPD with a RUNX1 p.P240Hfs mutation, the other was the index patient of a kindred with a novel RUNX1 variant, RUNX1 c.587C>T (p.T196I), noted on a targeted genetic testing of the B-ALL diagnostic sample. We discuss the clinical course, treatment approaches, and the outcome for the 2 patients. Additionally, we describe transient resolution of the mild thrombocytopenia and bleeding symptoms during therapy, as well as the finding of clonal hematopoiesis with a TET2 mutant clone in 1 of the patients. It is critical to consider testing for germline RUNX1 mutations in patients presenting with B-ALL who have a personal or family history of thrombocytopenia, bleeding symptoms, or RUNX1 variants identified on genetic testing at diagnosis.
Introduction
Germline RUNX1 mutations underlie the autosomal-dominant familial platelet disorder with associated myeloid malignancy (OMIM #601399), also known as RUNX1-familial platelet disorder (RUNX1-FPD).1 Patients with RUNX1-FPD may have qualitative and/or quantitative defects in platelets manifesting as bleeding symptoms.1 With a median onset at 29 years of age,2 the most common types of hematologic malignancies are myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), with rare diagnoses of T-cell acute lymphoblastic leukemia and lymphoma.2,3 Only 1 case of B-cell acute lymphoblastic leukemia (B-ALL) has been reported associated with a RUNX1 p.G217R mutation, which was not confirmed to be germline.4RUNX1 p.G217R variant in isoform RUNX1c (p.G190R in RUNX1b as reported by the investigators, ClinVarID 436614) is present at a low frequency in population databases (0.0031%, 9 alleles in gnomAD; PM2 and PS4 not met), and computational algorithms do not provide sufficient evidence to predict the pathogenicity of this variant (REVEL = 0.624; PP3 and BP4 not met). Additionally, this variant is not known to be located in a conserved and well-established functional domain or mutational hot spot (PM1 not met).
Here, we report the first 2 cases of B-ALL in patients with confirmed germline RUNX1 mutations.
Case description
The first patient was brought to medical attention as a 2-year-old with easy bruising and frequent epistaxis. She had mild thrombocytopenia (platelet count 90 000-130 000 cells per microliter) and normal prothrombin time, partial thromboplastin time, von Willebrand factor, ristocetin cofactor activity, and factor VIII levels. Ristocetin-induced platelet aggregation titration assays were abnormal on 2 occasions, leading to the presumptive diagnosis of type 2B von Willebrand disease, which was later ruled out with negative genetic testing. Her bleeding symptoms included bruising, epistaxis, menorrhagia, and excessive bleeding after loss of primary teeth requiring administration of antihemophilic factor/von Willebrand factor complex. She presented at the age of 16 years with a 2-month history of frequent febrile illnesses and pancytopenia; the diagnosis of B-ALL was made including flow cytometric confirmation of lineage. Flow cytometric analysis showed a blast population coexpressing CD45dim, CD19, CD34, terminal deoxynucleotidyl transferase, CD10 (subset), CD117 (small subset), CD22dim/−, cytosplasmic CD79a, CD38, CD58, CD81, and HLA-DR but lacking expression of CD20, CRLF2, surface immunoglobulins, and myelomonocytic (CD13, CD15, CD33) and T-cell (CD3, CD5, CD7, CD56) markers. Cytogenetics showed an abnormal karyotype (47,XX,+8), which has been reported previously but is rare in B-ALL.5 Targeted next-generation sequencing (Rapid Heme Panel6) of the diagnostic bone marrow sample revealed the following mutations: RUNX1C (c.587C>T, p.T196I) and TET2 (c.2399_2400delAT, p.H800Qfs*15) with variant allelic fractions (VAFs) of 50.3% and 4%, respectively. The patient was enrolled in the open trial at Dana-Farber Cancer Institute for treatment of B-ALL, which was approved by the Institutional Review Board of Dana-Farber Cancer Institute. She achieved remission after a 4-drug induction therapy (dexamethasone, vincristine, pegaspargase, doxorubicin) but had high minimal residual disease (MRD) at day 33 of treatment (4.2 × 10−3 clonal cells by clonoSEQ). Per protocol guidelines, she was treated with intensified therapy, resulting in low MRD at day 83 (8.8 × 10−4 clonal cells). She completed the full treatment in the next 24 months and tolerated it well, without delays or complications. Surprisingly, her platelet count at the start of each cycle (150 000-240 000 cells per microliter) was considerably higher than her baseline count prior to the leukemia diagnosis. Upon completion of therapy, her mild thrombocytopenia and bleeding symptoms returned. The patient remains in remission after 34 months.
The second patient has a family history of RUNX1-FPD with confirmed germline RUNX1 p.P240Hfs mutation (ClinVarID 561255).7 The patient’s mother has RUNX1-FPD with mild thrombocytopenia and developed MDS at the age of 32 years. The patient’s 3 siblings also have RUNX1-FPD, 2 with thrombocytopenia and petechiae. The patient remained asymptomatic until the age of 16 years, when he presented with petechiae, a platelet count of 11 000 cells per microliter, a white blood cell count of 54 000 cells per deciliter, and peripheral blasts, and was diagnosed with B-ALL, including flow cytometric confirmation of lineage. Flow cytometric analysis showed a blast population coexpressing CD45low, CD19, CD10, CD34, CD20, CD9, CD38, CD58, CD22, nuclear terminal deoxynucleotidyl transferase, and cytoplasmic CD79a, and negative for myeloperoxidase and cytoplasmic and surface CD3, CD5, CD13, and CD33. Cytogenetics showed a normal karyotype. Next-generation sequencing (FoundationOne Heme) of the bone marrow sample demonstrated an EBF1-PDGFRB fusion, a recurrent lesion in Ph-like B-ALL and associated with poor prognosis.8 He was treated per the Children’s Oncology Group B-ALL regimen with a 4-drug induction (prednisone, vincristine, pegaspargase, daunorubicin), with dasatinib added at midinduction and continued through consolidation. The Children’s Oncology Group AALL1732 trial was approved by the Institutional Review Board of the University of Alabama. Remission was achieved, with high MRD after the induction cycle and negative by flow cytometry after consolidation therapy (cyclophosphamide, cytarabine, 6-mercaptopurine, vincristine, pegaspargase, and dasatinib). His platelet count after the consolidation cycle was ∼300 000 cells per microliter, higher than his baseline level (152 000-174 000 cells per microliter) prior to the diagnosis of leukemia. Given his known RUNX1-FPD–associated predisposition to developing hematologic malignancies, in the setting of a high MRD at the end of induction, for a B-ALL with a genetic lesion associated with poor prognosis, he proceeded to haploidentical paternal-donor hematopoietic stem cell transplantation (HSCT) with posttransplantation cyclophosphamide, following fludarabine and total body irradiation–based conditioning. He is 1 year post-HSCT without significant transplantation-related complications.
Methods
With the history of chronic thrombocytopenia and bleeding symptoms in patient 1, the RUNX1 c.587C>T variant at a VAF of 50% led to the suspicion of it being a germline mutation. This was confirmed by the presence of the mutation in the patient’s fibroblasts and in the peripheral blood of her father, who also had lifelong thrombocytopenia and bleeding symptoms. We classified RUNX1 c.587C>T (p.T196I) as “likely pathogenic.”9 It is absent from population databases (PM2), it is found in a proband with a personal and family history of RUNX1-FPD, representing an increase over the normal population (PS4-supporting), computational predictions support a deleterious effect on gene function (REVEL = 0.967; PS3), it is located in a conserved mutation hotspot of RUNT homology domain (PM1), and additional variants have been identified at this position (p.T196R, p.T196A, PM5).
Results and discussion
RUNX1 p.T196I has not previously been reported in RUNX1-FPD; however, other amino acid substitutions at codon 196 have been described. Variants p.T196R10,11 and p.T196X12 have been associated with myeloid malignancies, and p.T196A13 has been associated with thrombocytopenia without reported malignancy. Lymphoid malignancies, most commonly T-cell acute lymphoblastic leukemia, have been reported in RUNX1-FPD (Figure 1), although no germline mutation appears to specifically predispose to only lymphoid malignancies; most families present with a mixture of lymphoid and myeloid malignancies.3,14 The low incidence of B-ALL in RUNX1-FPD is unusual. The ETV6-RUNX1 translocation, which conserves the RUNX1 protein in almost its entirety,14 is the most common genetic aberration in childhood B-ALL.15 Studies of murine Runx1 deficiency have suggested a critical role for RUNX1 in the maintenance of early pre–B-cell progenitors, where its loss induces a block in B-cell differentiation.16 The fusion to ETV6 could direct the RUNX1 protein to new targets, effectively resulting in apparent RUNX1 deficiency on certain loci.16 Further studies are needed to explore the possible common mechanism by which ETV6-RUNX1 and RUNX1 deficiency may lead to B-cell leukemogenesis.
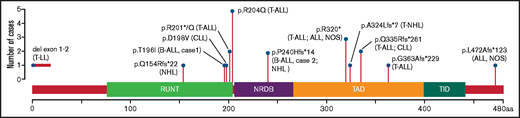
Schematic representation of RUNX1 showing the germline genetic variants in RUNX1-FPD associated with lymphoid malignancies. Shown are RUNX1 single-nucleotide variants and a deletion (del) reported in this study and reported by Brown et al,3 associated with lymphoid malignancies, annotated to RUNX1C; NM_001754; Ensembl: ENST00000300305. aa, amino acids; ALL, NOS, acute lymphoblastic leukemia, not otherwise specified; CLL, chronic lymphocytic leukemia; NHL, non-Hodgkin lymphoma; T-ALL, T-cell acute lymphoblastic leukemia; T-LL, T lymphoblastic lymphoma; T-NHL, T-cell non-Hodgkin lymphoma; NRDB, negative regulatory domain for DNA binding; TAD, transcriptional activation domain; TID, transcriptional inhibitory domain.
Schematic representation of RUNX1 showing the germline genetic variants in RUNX1-FPD associated with lymphoid malignancies. Shown are RUNX1 single-nucleotide variants and a deletion (del) reported in this study and reported by Brown et al,3 associated with lymphoid malignancies, annotated to RUNX1C; NM_001754; Ensembl: ENST00000300305. aa, amino acids; ALL, NOS, acute lymphoblastic leukemia, not otherwise specified; CLL, chronic lymphocytic leukemia; NHL, non-Hodgkin lymphoma; T-ALL, T-cell acute lymphoblastic leukemia; T-LL, T lymphoblastic lymphoma; T-NHL, T-cell non-Hodgkin lymphoma; NRDB, negative regulatory domain for DNA binding; TAD, transcriptional activation domain; TID, transcriptional inhibitory domain.
Both patients tolerated their respective chemotherapy without major toxicity. Patient 1 experienced improved platelet counts and had resolution of long-standing bleeding symptoms throughout therapy, which recurred upon completion of chemotherapy. Patient 2 also had platelet counts that were higher than his pre–B-ALL baseline levels after he achieved MRD-negative status. In preclinical models, stress thrombopoiesis with chemotherapy has been shown to be mediated through TAL1 and its target NF-E2.17 NF-E2 and RUNX1 co-occupy cis elements in megakaryocytes for critical activity of genes in thrombopoiesis.18 It is not known whether RUNX1 deficiency potentiates stress thrombopoiesis, whether stress thrombopoiesis overcomes RUNX1-FPD–related thrombocytopenia, or whether certain cytotoxic drugs enhance the activity or the abundance of the wild-type RUNX1 protein, similar to proteosome inhibitors,19 temporarily correcting the thrombocytopenia.
Patient 1 was 16 years old at the time of B-ALL diagnosis and was found to have an acquired mutation in TET2 at a 4% VAF. Even though the clone with trisomy 8 in the diagnostic marrow disappeared at remission, the TET2 mutant clone persisted through therapy with a 5.5% VAF in peripheral blood 10 months after the end of therapy. Mutations in TET2 are one of the most common mutations underlying clonal hematopoiesis of indeterminate potential in adults.20 Although exceedingly rare in children, it has been described in young patients with RUNX1-FPD.3,21 Ongoing research on serial assessment of mutant clones over time and association with disease diagnosis will be critical for our understanding of specific risks that mutant clones in clonal hematopoiesis or their rate of growth carry in individuals with germline predisposition mutations.
Lastly, we wanted to address the difference in the approach of B-ALL therapy for the 2 patients. In the absence of a high-risk genomic alteration, patient 1 proceeded to completion of acute lymphoblastic leukemia–directed treatment upon achieving low MRD status after intensification therapy. Patient 2 proceeded to HSCT for the treatment of B-ALL, as well as eradication of his future leukemia risk associated with RUNX1-FPD, in the setting of the diagnosis of B-ALL with a poor-prognostic genetic fusion and an available haploidentical related donor. Patients with RUNX1-FPD have a high lifetime risk of developing MDS and AML, but it is unclear whether this risk is increased further with the use of cytotoxic chemotherapy, such as those in B-ALL treatment regimens that modestly increase the risk of secondary hematologic malignancy in survivors. The recommendation for a preventative HSCT, as well as treatment of a high-risk B-ALL in case 2, remains a highly complex one in the field of predisposition genetics in pediatrics.22 With growing evidence placing RUNX factors in the response pathways to DNA damage,23 future work will be needed to understand the role that acute lymphoblastic leukemia–directed cytotoxic chemotherapy may play in changing the risk for the potential development of secondary leukemias in RUNX1-FPD.
One should consider an underlying RUNX1-FPD syndrome in patients with a diagnosis of B-ALL who have personal or family history of thrombocytopenia or bleeding symptoms and/or who have RUNX1 variants identified on next-generation sequencing platforms in a diagnostic sample. The diagnosis of a germline predisposition syndrome has profound implications for the patient, the management of their diagnosis, and future monitoring, as well as for family members.
Acknowledgments
This work was supported by RUNX1 Research Program and Alex’s Lemonade Stand Foundation (A.L.B. and A.B.C.).
Authorship
Contribution: K.A.S., U.G. and S.A. wrote the manuscript; A.L.B. completed genomic analyses; A.E.P., A.B.C., M.A.K. and S.A. interpreted the data and revised the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Serine Avagyan, Dana-Farber/Boston Children’s Hospital Cancer and Blood Disorders Center, 450 Brookline Ave, Boston, MA 02215; e-mail: serine_avagyan@dfci.harvard.edu.

