

Key Points
GATA2 mutations are more common than expected in patients with myeloid malignancy who develop invasive aspergillosis.
Myeloid malignancy patients with somatic GATA2 mutations have a high risk of invasive fungal disease with antineoplastic treatment.

Abstract
Invasive fungal disease (IFD) can be a severe treatment complication in patients with myeloid malignancies, but current risk models do not incorporate disease-specific factors, such as somatic gene mutations. Germline GATA2 deficiency is associated with a susceptibility to IFD. To determine whether myeloid gene mutations were associated with IFD risk, we identified 2 complementary cohorts of patients with myeloid malignancy, based on (1) the diagnosis of invasive aspergillosis (IA), or (2) the presence of GATA2 mutations identified during standard clinical sequencing. We found somatic GATA2 mutations in 5 of 27 consecutive patients who had myeloid malignancy and developed IA. Among 51 consecutive patients with GATA2 mutations identified in the evaluation of myeloid malignancy, we found that IFD was diagnosed and treated in 21 (41%), all of whom had received chemotherapy or had undergone an allogeneic stem cell transplant. Pulmonary infections and disseminated candidiasis were most common. The 90-day mortality was 52% among patients with IFD. Our results indicate that patients with somatic GATA2 mutations are a vulnerable subgroup of patients with myeloid malignancy who have high risk for treatment-associated IFD and suggest that a focused approach to antifungal prophylaxis be considered.
Introduction
The GATA2 gene encodes a transcription factor and is mutated recurrently in sporadic myeloid malignancies and in patients with GATA2 deficiency syndrome.1-5 Missense mutations affecting the GATA2 zinc finger DNA-binding domains (ZF1 and ZF2) cause loss of function byimpairing binding to GATA-DNA motifs, whereas nonsense or frameshift mutations reduce the overall abundance of GATA2 protein.4 Patients with germline GATA2 mutations can have a primary immunodeficiency and an elevated risk of recurrent infections that are, in part, caused by myeloid dendritic cell, monocyte, and natural killer cell dysfunction.4-6 These defects in immune cells confer susceptibility to invasive fungal disease (IFD), particularly invasive aspergillosis (IA).7,8
GATA2 is mutated somatically in 1% to 4% of patients with sporadic myeloid malignancies.1-3 One study found that 5 of 9 patients with myeloid malignancies and somatic GATA2 mutations had clinical and flow cytometric features of immunodeficiency, suggesting that somatic mutations exert pleiotropic effects in terminal immune lineages in addition to their effects on hematopoietic stem and progenitor cells.9 A case report identified a patient with a somatic GATA2-mutated myeloid neoplasm who developed mycobacterial and invasive pulmonary fungal infections.10 These reports raise the possibility that somatic GATA2 deficiency confers a risk of infection similar to that of germline mutations. We therefore sought to define the prevalence and spectrum of IFD in a consecutive series of adult patients with myeloid malignancy who harbored somatic GATA2 mutations.
Study design
Genetic analysis
IA cohort
Twenty-seven consecutive adult patients had clinical sequencing in an evaluation of myeloid malignancy and were diagnosed with proven or probable IA from March 2015 through December 2017 (Figure 1A).13
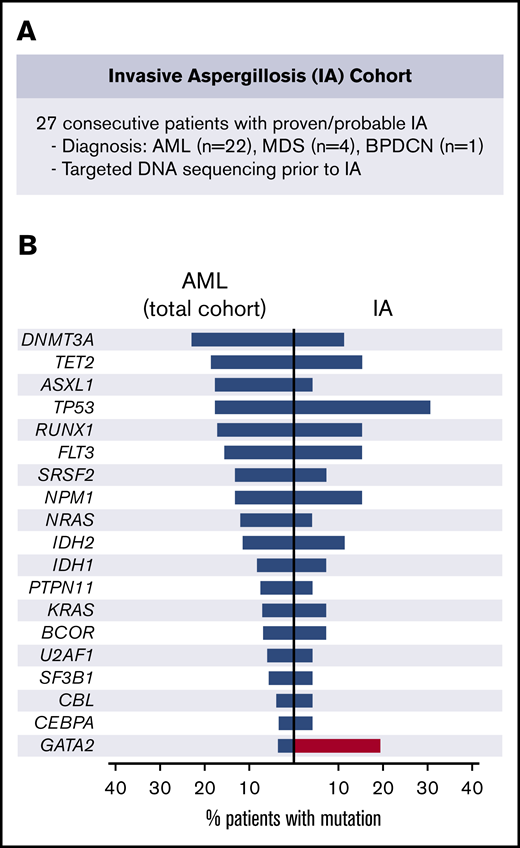
IA cohort. (A) Description of the IA cohort. (B) Frequency of recurrent driver mutations in patients with AML at our institution (left) and observed frequency in the IA cohort (right). BPDCN, blastic plasmacytoid dendritic cell neoplasm.
IA cohort. (A) Description of the IA cohort. (B) Frequency of recurrent driver mutations in patients with AML at our institution (left) and observed frequency in the IA cohort (right). BPDCN, blastic plasmacytoid dendritic cell neoplasm.
GATA2 cohort
Seventy-six patients with GATA2 mutations were identified among 2383 consecutive adult patients who had clinical sequencing for evaluation of acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), or MDS/MPN overlap from July 2014 through September 2019. Seven patients with proven germline GATA2 mutations were excluded from the analysis. Fifty-one patients were considered evaluable for IFD based on (1) documentation of death at any time, or (2) at least 90 days of follow-up after identification of GATA2 mutation by sequencing. In the remaining 51 patients, GATA2 mutations were defined as either somatic or likely somatic based on genetic characteristics and clinical history obtained by a detailed review of personal and family history of myeloid malignancies, immunodeficiency, and recurrent infections (Figure 2A).
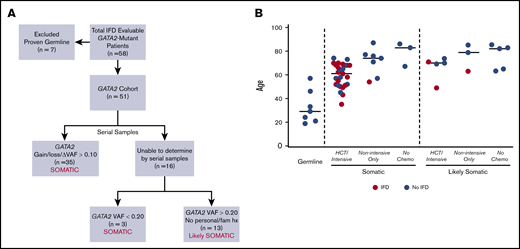
GATA2 cohort. (A) The approach to defining GATA2 mutations as somatic or likely somatic. (B) Age distribution comparing patients with proven germline GATA2 mutations (n = 7) and patients with definitive (n = 38) or likely (n = 13) GATA2 mutations, based on the framework outlined in panel A. Red points indicate proven, probable, or possible-treated cases of IFD. VAF, variant allele frequency.
GATA2 cohort. (A) The approach to defining GATA2 mutations as somatic or likely somatic. (B) Age distribution comparing patients with proven germline GATA2 mutations (n = 7) and patients with definitive (n = 38) or likely (n = 13) GATA2 mutations, based on the framework outlined in panel A. Red points indicate proven, probable, or possible-treated cases of IFD. VAF, variant allele frequency.
Annotation of fungal disease
IA and IFD were categorized as proven, probable, or possible based on the revised guidelines from the European Organization for the Research and Treatment of Cancer and Mycoses Study Group (EORTC/MSG).14 Patients with possible IFD were separated into those who received antifungal agents (possible-treated) and those who did not. See supplemental Methods for further details. Based on the low baseline rate of IFD, antifungal prophylaxis during treatment of myeloid malignancies is not standard practice at our institution. This study was conducted with the approval of the Dana-Farber/Harvard Cancer Center and Massachusetts General-Brigham and Women’s Institutional Review Boards.
Results and discussion
To identify potential associations between myeloid genetic alterations and risk of IFD, we first analyzed 27 consecutive patients with myeloid malignancies who developed IA (supplemental Table 1) and compared the frequency of somatic gene mutations in this cohort to the frequency of myeloid mutations in patients with AML and MDS at our institution (supplemental Table 2). The most commonly mutated genes in patients with IA were TP53 (29%; 8 of 27) and GATA2 (19%; 5 of 27), in contrast with their frequencies in patients with AML in the overall cohort of 17.3% and 3.2%, respectively (Figure 1B; supplemental Table 2). TP53 and GATA2 were not concurrently mutated in individual patients, indicating that they reflect independent markers of IA risk. Whereas TP53 mutations are associated with factors previously linked to increased risk of IA, such as adverse cytogenetics and treatment resistance,15,16 GATA2 mutations, typically in the context of normal karyotype AML, have had no adverse impact on outcomes.17 In this cohort, the number of lines of prior therapy was not significantly different between the GATA2 mutant and wild-type cases (Mann-Whitney, P = .25).
Germline GATA2 mutations have been linked to an increased risk of fungal infections,4,6-8 raising the possibility that somatic GATA2 mutations mediate a similar effect. To test this hypothesis, we determined the incidence of IFD in a larger cohort of consecutive patients with GATA2 mutations. Among 51 evaluable patients with GATA2 mutations, diagnoses included AML (n = 27), MDS (n = 8), MDS/MPN overlap syndromes (n = 13), and MPN (n = 3). Most of these patients (43 of 51; 84%) received myelotoxic treatments, including intensive induction chemotherapy, hypomethylating agents, and allogeneic hematopoietic cell transplantation (HCT) (supplemental Tables 3 and 4). Consistent with the reported spectrum of pathogenic GATA2 mutations,4 the mutations in our cohort included truncating mutations located throughout the coding region and missense substitutions or in-frame indels located within ZF1 or ZF2 (supplemental Figure 2).
In total, 21 of 51 (41%) evaluable patients with GATA2 mutations developed proven (n = 5), probable (n = 10), or possible-treated (n = 6) IFD (Table 1). Of the 21 patients, 18 had definitively somatic and 3 had likely somatic GATA2 mutations (Figure 2B). There was no significant association between the diagnosis of IFD and the type, zinc finger distribution, or variant allele frequency of the GATA2 mutation or the presence of specific cooccurring gene mutations (supplemental Table 5; supplemental Figure 3). In all 21 patients with proven, probable, or possible-treated IFD, the development of IFD followed treatment with chemotherapy or HCT (Figures 2B and 3; Table 1). By contrast, there were no cases of IFD in the 8 patients with GATA2-mutated disease who did not receive chemotherapy or undergo HCT, because of their lower risk disease or fitness, despite similar follow-up periods. IFD diagnoses occurred evenly across the 5-year study period, with no temporal clustering of cases suggestive of environmental outbreaks (supplemental Figure 4).
Clinical, genetic, and IFD characteristics in the GATA2 Cohort
| Patient ID | Age, y | Disease/treatment | GATA2 | IFD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | HCT | Proximal chemotherapy | Mutation | VAF | Somatic confidence | Category | Infection site | Mycology | ANC <0.5 × 103/μL | ||
| G1 | 51 | AML | Yes | MEC | N317H | 0.12 | Definite | Proven | Brain | Pathology with invasive fungal forms, culture with Scedosporium sp. | Yes |
| G2 | 68 | AML | No | MEC | R398W | 0.30 | Definite | Proven | Pulmonary, disseminated candidiasis | Elevated serum galactomannan, C tropicalis bloodstream infection | Yes |
| G3 | 58 | MDS | Yes | — | R398W | 0.37 | Definite | Proven | Pulmonary, disseminated candidiasis | Elevated serum galactomannan, C albicans bloodstream infection | Yes |
| G4 | 72 | AML | Yes | — | G320D | 0.37 | Likely | Proven | Disseminated candidiasis | C tropicalis bloodstream infection | Yes |
| G5 | 51 | MDS/MPN | Yes | — | R396Q | 0.08 | Definite | Proven | Pulmonary | Pathology with invasive fungal forms, BAL culture with Rhizopus sp. | No |
| G6 | 49 | AML | Yes | Decitabine | G320D | 0.44 | Definite | Probable | Sinus | Elevated serum galactomannan | Yes |
| G7 | 70 | AML | Yes | Clofarabine, cytarabine | T301K | 0.31 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G8 | 35 | AML | Yes | Decitabine | L321P | 0.16 | Definite | Probable | Pulmonary | BAL culture with A fumigatus | Yes |
| G9 | 53 | AML | Yes | MEC | A12fs* | 0.44 | Definite | Probable | Pulmonary | Elevated BAL galactomannan | Yes |
| G10 | 68 | AML | No | CPX-351 | M388_K390del | 0.31 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G11 | 63 | AML | No | Decitabine/venetoclax | G320D | 0.41 | Likely | Probable | Pulmonary | Elevated serum galactomannan | Yes |
| G12 | 61 | MDS | No | CPX-351 | G82fs* | 0.08 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G13 | 50 | AML | Yes | — | N297S | 0.45 | Likely | Probable | Pulmonary | Elevated serum galactomannan/β-d-glucan | Yes |
| G14 | 68 | MDS/MPN | Yes | Decitabine | P385R | 0.13 | Definite | Probable | Pulmonary | BAL culture with A fumigatus | No |
| G15 | 62 | AML | Yes | — | V16fs* | 0.38 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G16 | 54 | AML | No | αCD123 ADC | K282fs* | 0.21 | Definite | Possible-treated | Pulmonary | — | Yes |
| G17 | 66 | AML | No | Daunorubicin, cytarabine | T354M, P121fs* | 0.44, 0.36 | Definite | Possible-treated | Pulmonary | — | Yes |
| G18 | 66 | AML | No | Daunorubicin, cytarabine | D99fs* | 0.18 | Definite | Possible-treated | Pulmonary | — | Yes |
| G19 | 70 | AML | No | MEC | M388_E391del | 0.44 | Definite | Possible-treated | Pulmonary | — | Yes |
| G20 | 55 | AML | No | Daunorubicin, cytarabine | G274fs* | 0.13 | Definite | Possible-treated | Pulmonary | — | Yes |
| G21 | 43 | MDS/MPN | Yes | — | L321F | 0.15 | Definite | Possible-treated | Pulmonary | — | Yes |
| Patient ID | Age, y | Disease/treatment | GATA2 | IFD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | HCT | Proximal chemotherapy | Mutation | VAF | Somatic confidence | Category | Infection site | Mycology | ANC <0.5 × 103/μL | ||
| G1 | 51 | AML | Yes | MEC | N317H | 0.12 | Definite | Proven | Brain | Pathology with invasive fungal forms, culture with Scedosporium sp. | Yes |
| G2 | 68 | AML | No | MEC | R398W | 0.30 | Definite | Proven | Pulmonary, disseminated candidiasis | Elevated serum galactomannan, C tropicalis bloodstream infection | Yes |
| G3 | 58 | MDS | Yes | — | R398W | 0.37 | Definite | Proven | Pulmonary, disseminated candidiasis | Elevated serum galactomannan, C albicans bloodstream infection | Yes |
| G4 | 72 | AML | Yes | — | G320D | 0.37 | Likely | Proven | Disseminated candidiasis | C tropicalis bloodstream infection | Yes |
| G5 | 51 | MDS/MPN | Yes | — | R396Q | 0.08 | Definite | Proven | Pulmonary | Pathology with invasive fungal forms, BAL culture with Rhizopus sp. | No |
| G6 | 49 | AML | Yes | Decitabine | G320D | 0.44 | Definite | Probable | Sinus | Elevated serum galactomannan | Yes |
| G7 | 70 | AML | Yes | Clofarabine, cytarabine | T301K | 0.31 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G8 | 35 | AML | Yes | Decitabine | L321P | 0.16 | Definite | Probable | Pulmonary | BAL culture with A fumigatus | Yes |
| G9 | 53 | AML | Yes | MEC | A12fs* | 0.44 | Definite | Probable | Pulmonary | Elevated BAL galactomannan | Yes |
| G10 | 68 | AML | No | CPX-351 | M388_K390del | 0.31 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G11 | 63 | AML | No | Decitabine/venetoclax | G320D | 0.41 | Likely | Probable | Pulmonary | Elevated serum galactomannan | Yes |
| G12 | 61 | MDS | No | CPX-351 | G82fs* | 0.08 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G13 | 50 | AML | Yes | — | N297S | 0.45 | Likely | Probable | Pulmonary | Elevated serum galactomannan/β-d-glucan | Yes |
| G14 | 68 | MDS/MPN | Yes | Decitabine | P385R | 0.13 | Definite | Probable | Pulmonary | BAL culture with A fumigatus | No |
| G15 | 62 | AML | Yes | — | V16fs* | 0.38 | Definite | Probable | Pulmonary | Elevated serum β-d-glucan | Yes |
| G16 | 54 | AML | No | αCD123 ADC | K282fs* | 0.21 | Definite | Possible-treated | Pulmonary | — | Yes |
| G17 | 66 | AML | No | Daunorubicin, cytarabine | T354M, P121fs* | 0.44, 0.36 | Definite | Possible-treated | Pulmonary | — | Yes |
| G18 | 66 | AML | No | Daunorubicin, cytarabine | D99fs* | 0.18 | Definite | Possible-treated | Pulmonary | — | Yes |
| G19 | 70 | AML | No | MEC | M388_E391del | 0.44 | Definite | Possible-treated | Pulmonary | — | Yes |
| G20 | 55 | AML | No | Daunorubicin, cytarabine | G274fs* | 0.13 | Definite | Possible-treated | Pulmonary | — | Yes |
| G21 | 43 | MDS/MPN | Yes | — | L321F | 0.15 | Definite | Possible-treated | Pulmonary | — | Yes |
αCD123-ADC, αCD123-targeting antibody-drug conjugate; ANC, absolute neutrophil count; MEC, mitoxantrone, etoposide, and intermediate-dose Ara-C; VAF, variant allele frequency (VAF at initial index detection).
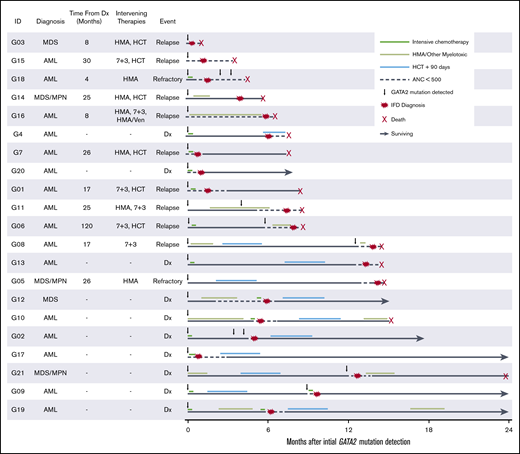
Disease, treatment, and IFD timeline. Swimmers plot detailing timeline of disease and treatment relative to initial GATA2 detection in patients with proven, probable, or possible-treated IFD in the GATA2 cohort. Dx, diagnosis; HMA, hypomethylating agent.
Disease, treatment, and IFD timeline. Swimmers plot detailing timeline of disease and treatment relative to initial GATA2 detection in patients with proven, probable, or possible-treated IFD in the GATA2 cohort. Dx, diagnosis; HMA, hypomethylating agent.
Pulmonary infection was the most common manifestation of IFD (18 of 21 patients). There was no evidence of preexisting pulmonary alveolar proteinosis, which is associated with germline GATA2 deficiency syndrome,18 in the patients with pulmonary IFD. Five patients with pulmonary consolidation had concurrently elevated serum or bronchoalveolar lavage (BAL) galactomannan levels. Disseminated candidiasis with positive blood culture isolates occurred in 3 patients. One patient developed invasive fungal sinusitis and another had a fungal brain abscess. Culture isolates included Aspergillus fumigatus, Rhizopus sp., Scedosporium sp., Candida albicans, and Candida tropicalis. All 6 patients with possible-treated IFD had concern for isolated pulmonary infection, including a patient with a BAL culture that grew Doratomyces sp. All 21 patients received treatment with mold-active antifungals, including triazoles or liposomal amphotericin B. The 90-day mortality after diagnosis of IFD was 52%. Complete descriptions of the clinical, genetic, and mycological characteristics of patients with IFD are shown in Table 1.
The depth and duration of neutropenia are established risk factors for development of IFD.15 Therefore, we evaluated the characteristics of neutropenia in this cohort to determine whether GATA2 mutations were associated with severe or prolonged treatment-induced neutropenia. As expected, most patients (19 of 21) were severely neutropenic (ANC <0.5 × 103/μL) at the time of the IFD diagnosis. In the subset of 13 patients with GATA2-mutated AML who received intensive induction chemotherapy at diagnosis or relapse, the median duration from the start of chemotherapy to ANC recovery (>0.5 × 103/μL) was 27 days. This was consistent with the median duration of severe neutropenia reported in a cohort of 205 consecutive patients with AML treated with 7+3 therapy (23 days; interquartile range, 19-28).19 Further, in our GATA2 cohort, there was no difference in the median duration of severe neutropenia between patients with AML who did and those who did not develop IFD after induction (28 vs 25 days; P = .67). Similarly, because patients with germline GATA2 deficiency can also have quantitative reduction in monocytes, we examined the absolute monocyte count (AMC) at the index time point. We found absolute monocytopenia (AMC <0.5 × 103/μL) in 31 of 51 patients (61%), but there was no difference in AMC between patients who did and those who did not develop IFD. Together, these observations suggest that the effect of GATA2 mutations on IFD risk is not primarily mediated by a selective quantitative defect in neutrophils or monocytes.
We found that 21 of 43 patients (49%) with GATA2-mutated myeloid malignancy who were treated with chemotherapy or HCT developed proven, probable, or possible IFD and received antifungal therapy. IFD developed in 10 of 26 (38%) treated patients with GATA2 mutations identified at diagnosis and in 11 of 17 (65%) patients with GATA2 mutation identified in the setting of relapsed or refractory disease. The high incidence of IFD in patients with GATA2 mutations contrasted sharply with the rate of IFD in patients with myeloid malignancy in multiple treatment contexts. Specifically, we observed a 6% cumulative incidence of IFD at 1 year in 901 patients with myeloid malignancy who underwent allogeneic HCT at our institution, and the incidence of IFD in patients with GATA2-mutated myeloid malignancy in our cohort was higher than reported in patients who received induction chemotherapy for acute leukemia (10% within 100 days and 13% overall),20 during treatment with hypomethylating agents (9.6%)21 and after HCT (10% within 1 year).22 These data indicate that the presence of somatic GATA2 mutations may define a specific subgroup of patients who have an elevated risk of developing IFD in the context of myelotoxic or immunosuppressive therapy.
Randomized, placebo-controlled trials have demonstrated a reduction in IFD in patients who undergo HCT and receive antifungal prophylaxis, compared with those with no prophylaxis after HCT.23,24 However, these studies evaluated patients in uniform disease cohorts, without the ability to analyze the magnitude of effect in genetically defined subpopulations. Recent studies have demonstrated that mutations in MDS/AML driver genes can exert pleiotropic effects in terminal hematopoietic lineages in addition to promoting clonal expansion of stem and progenitor cells. For example, the JAK2-V617F mutation has been linked with increased formation of neutrophil extracellular traps and increased thrombotic risk in myeloproliferative neoplasms,25 and TET2 mutations have been associated with potentiated immune/inflammatory signaling and cardiovascular risk in individuals with clonal hematopoiesis.26,27 Our results suggest that acquired GATA2 deficiency causes immune alterations that are similar to those defined in patients with germline GATA2 deficiency, thereby exposing them to a similarly increased risk of developing IFD. This increased risk appears to manifest particularly in the context of myelosuppressive chemotherapy and HCT, but not in the absence of treatment, suggesting that GATA2-dependent fungal conidial surveillance by mature innate immune cells suppresses overt IFD in the absence of treatment-related myelosuppression. Our findings indicate that somatic GATA2 mutations define a vulnerable subgroup of patients with myeloid malignancy for whom antifungal agents should be considered to be a part of the infection prophylaxis regimen.
Deidentified data are included in the supplemental material.
Acknowledgment
The authors thank the Dana-Farber Cancer Institute (DFCI) Hematologic Malignancies Data Repository (HMDR) for assistance in this study.
This work was supported by National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute fellowship training grant 2T32HL116324-06 (R.S.V.), NIH/National Institute of Allergy and Infectious Diseases fellowship training grant T32 AI007061-39 (T.D.B.), NIH/National Cancer Institute grant K08CA204734 (R.C.L.), and by the Dresner Foundation (R.C.L.).
Authorship
Contribution: R.S.V., M.P.C., F.M.M., T.D.B., and R.C.L designed the study; R.S.V., M.P.C., C.E.R., D.F., V.T.H., S.K., and T.D.B. performed the data analysis; R.S.V., T.D.B., and R.C.L. wrote the manuscript; and all authors reviewed the manuscript during its preparation and approved the submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: R. Coleman Lindsley, Dana-Farber Cancer Institute, 450 Brookline Ave, DA-530C, Boston, MA 02215; e-mail: coleman_lindsley@dfci.harvard.edu.

