

Abstract
Luspatercept is an activin receptor ligand trap that has been shown to enhance late-stage erythropoiesis in animal models of β-thalassemia. A multicenter, international, phase 2 dose-finding study was initiated in adult patients with β-thalassemia, either non–transfusion-dependent thalassemia (NTDT) or transfusion-dependent thalassemia (TDT). Positive results of the phase 2 study paved the way to a randomized phase 3 clinical trial (BELIEVE) to assess the efficacy and safety of luspatercept. The BELIEVE trial is a randomized, double-blind, placebo-controlled phase 3 trial. Three hundred thirty-six patients aged ≥18 years with TDT (regularly transfused, 6-20 red blood cell units within 24 weeks before randomization) were included in the trial. Patients received luspatercept or placebo subcutaneously every 21 days for ≥48 weeks and best supportive care. Forty-eight of 224 patients (21.4%) in the luspatercept group achieved the primary end points (≥33% reduction in transfusion burden) compared with those in the placebo group (4.5%; P < .001). Moreover, more patients had a ≥33% reduction in transfusion burden during any rolling 12-week interval (70.5% vs 29.5%) or any 24-week interval (41.1% vs 2.7%) with luspatercept than with the placebo. Transfusion independence was achieved by 11% of patients in the luspatercept group. Transient adverse events were more frequent with luspatercept than with placebo, but were manageable. Luspatercept was approved by the US Food and Drug Administration in 2019 and by the European Medicines Agency in 2020. The luspatercept trial is registered on www.clinicaltrials.gov at #NCT01749540 and the BELIEVE trial at #NCT02604433.
Introduction
Thalassemia syndromes are inherited hemoglobinopathies characterized by impaired or absent production of one of the globin chains of adult hemoglobin with subsequent accumulation of the unpaired chains. The most common form is β-thalassemia related to a defective production of the β-globin chains causing an unbalanced ratio of α-globin to β-globin. As a consequence, the unbound free α-globin chains precipitate in erythroid precursors, leading to ineffective erythropoiesis, chronic hemolytic anemia, and compensatory hemopoietic expansion.1 Ineffective erythropoiesis is the leading driver of clinical severity of β-thalassemias and, for many years, the standard of care to suppress it was red blood cell (RBC) transfusions.2 Bone marrow transplantation was introduced in the 1980s with the rationale of restoring the capability of producing functional hemoglobin.3 Gene therapy with globin lentiviral vectors and genome editing to inhibit the BCL11A gene are currently under investigation.4 These approaches, however, have several limitations, are feasible in a small subset of patients, and require transplantation conditioning. Thus, much effort has been devoted to finding new therapeutic options.
Luspatercept (formerly ACE-536; Acceleron Pharma, Celgene/Bristol Myers Squibb) is a ligand trap consisting of a modified form of the extracellular domain of the human activin type 2B receptor (ActR2B) linked to the fragment crystallizable (Fc) domain of human immunoglobulin G1, which binds to select transforming growth factor-β superfamily (TGF-β) ligands, blocks SMAD-2/3 signaling, and enhances erythroid maturation. Luspatercept was approved by the US Food and Drug Administration (FDA)5 in 2019 and by the European Medicines Agency6 in 2020 to treat anemia in adult patients with β-thalassemia who require regular RBC transfusions.
Luspatercept pharmacology
Members of the TGF-β superfamily ligands, which include TGF-β, activins, growth differentiation factors (GDFs), and bone morphogenetic proteins, have been shown to act as inhibitors of late-stage erythropoiesis.7,8 The activin receptor ligand traps sotatercept and luspatercept were designed to compete with the extracellular domains of activin receptor type 2A (ACVR2A) or 2B, to act as ligand traps for TGF-β–like molecules9 (Figure 1). Sotatercept was originally developed to treat bone loss disorders, but clinical studies unexpectedly revealed increased hematocrit and hemoglobin (Hb) levels in treated patients.10 Further studies showed that sotatercept and luspatercept, through their ability to reduce Smad-2/3 signaling, improve anemia in disorders characterized by ineffective erythropoiesis, such as β-thalassemia11 and myelodysplastic syndromes (MDSs).12 Luspatercept was tested in the Hbbth1/th1 transgenic mouse model of human β-thalassemia intermedia,11 which demonstrated an increase in RBC count, hematocrit, and Hb levels in a dose-dependent manner.13 Several studies in mouse models of anemia associated with ineffective erythropoiesis confirmed that Smad-2/3 and GDF-11 inhibition restores normal erythropoietic differentiation and improves anemia.14 On the basis of this observation, it was postulated that GDF-11 could be the target of luspatercept; however, in 2019, Guerra and colleagues15 clearly demonstrated that GDF-11 is not the only target of luspatercept; this observation was supported in a commentary by Camaschella.16 Subsequent to this observation, Martinez et al17 published consistent data showing that luspatercept enhances erythroid differentiation in murine β-thalassemia by increasing GATA-1 availability. Using differentiating murine erythroleukemia cells and GDF-11–induced overactivation of the Smad-2/3 pathway, they found a higher nuclear localization of pSmad-2/3 and Smad-4 and a concomitant reduced nuclear localization and expression of GATA-1 and TIF-1. In addition, ACE-536 (luspatercept) increased the nuclear localization of TIF1-γ and the expression of GATA-1.17 A consistent body of data has demonstrated that overactivation of the Smad-2/3 signaling pathway negatively regulates terminal erythroid differentiation in mouse models of β-thalassemia, partly by reducing GATA-1 expression. RAP-536 (murine ACE-536/luspatercept)-mediated inhibition of Smad-2/3 signaling enhances erythroid maturation in this context by increasing the expression and functional availability of GATA-1. It is well known that GATA-1 is indispensable for erythroid maturation.18,19
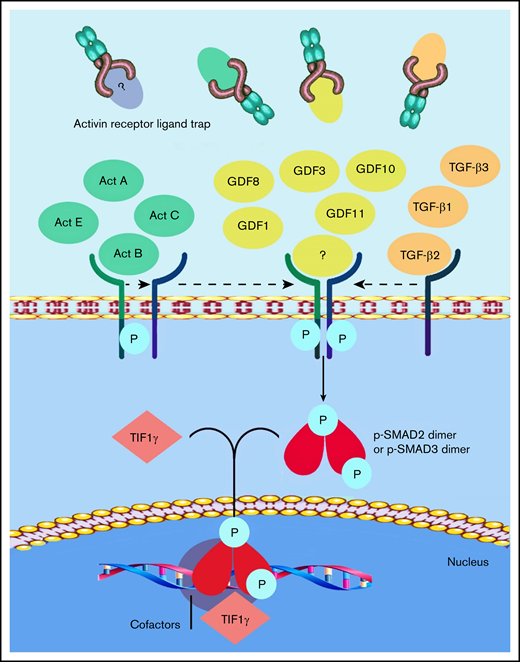
Current understanding of the mechanism of action of luspatercept. The SMAD-2/3 signaling pathway is the target of activin receptor ligand trap molecules. Members of the TGF-β superfamily ligand binding lead to the multimerization of type 1 and 2 receptors. Upon the activation of the type 1 receptor, phosphorylation of SMAD-2 and SMAD-3 takes place. Luspatercept prevents the binding of the ligand, thus inhibiting this pathway and promoting late-stage erythropoiesis.
Current understanding of the mechanism of action of luspatercept. The SMAD-2/3 signaling pathway is the target of activin receptor ligand trap molecules. Members of the TGF-β superfamily ligand binding lead to the multimerization of type 1 and 2 receptors. Upon the activation of the type 1 receptor, phosphorylation of SMAD-2 and SMAD-3 takes place. Luspatercept prevents the binding of the ligand, thus inhibiting this pathway and promoting late-stage erythropoiesis.
A recent article by Chen et al20 showed that the mean elimination half-life of luspatercept is ∼11 days.
Clinical development
The first study of luspatercept in humans was conducted in healthy volunteers who received 2 subcutaneous doses of either luspatercept (0.0625-0.25 mg/kg) or placebo every 2 weeks. Dose-dependent increases in Hb concentration were observed from 7 days after initiation of treatment and were maintained for several weeks. Eighty-three percent of the participants had up to a 1.0-g/dL Hb level increase in a dose-dependent manner. The drug was well tolerated, and no serious adverse events were observed.21 A multicenter, international, phase 2 dose-finding study was then initiated in adult patients with β-thalassemia, either non–transfusion-dependent (NTDT) or transfusion-dependent thalassemia (TDT). Primary end points of the study included erythroid response, defined as a Hb level increase from a baseline of ≥1.5 g/dL for ≥2 weeks (in the absence of RBC transfusions) for patients with NTDT and a reduction in RBC transfusion burden over a 12-week interval of ≥20%, compared with pretreatment for patients with TDT. Luspatercept was administered subcutaneously every 3 weeks at doses ranging from 0.2 to 1.25 mg/kg. In the NTDT group (n = 33), 58% of the patients achieved a mean Hb level increase from baseline of ≥1.5 g/dL over 14 consecutive days (95% confidence interval [CI], 39.1-75.5). The 6-minute walk test showed a significant improvement at 48 weeks of treatment, and improvement correlated with the Hb level.22 In the TDT group (n = 31), 81% achieved a transfusion burden reduction of ≥20% over any 12 weeks during the study, compared with the 12 weeks before baseline (95% CI, 63.6-92.8). In the treated patients overall, the adverse effects were tolerable, being grades 1 to 2: bone pain was seen in 38% of patients, headache in 28%, myalgia in 22%, and arthralgia in 19%. This study is now in a 5-year extension phase (www.clinicaltrials.gov; #NCT02268409).22
These findings, including achievement of the secondary end points, paved the way to a randomized phase 3 clinical trial (BELIEVE) to assess the efficacy and safety of luspatercept (all trial phases are shown in Table 1). Similar results were obtained with sotatercept23 ; however, luspatercept was selected for phase 3 development, mainly because, unlike sotatercept, it binds only minimally to activin A and thus may have lower off-target effects.
Sotatercept/luspatercept trials for β-thalassemia
| Trial identifier | Phase/status | Enrolled patients | Drug product | Sponsor/center |
|---|---|---|---|---|
| NCT01571635 | Phase 2; active, not recruiting | 16 TDT, 30 NTDT | Sotatercept (ACE-011) | Celgene; multicenter international sites |
| NCT01749540 | Phase 2; completed | 31 TDT, 33 NTDT | Luspatercept (ACE-536) | Acceleron Pharma; multicenter international sites |
| NCT02604433 | Phase 3; completed, not recruiting | 336 TDT | Luspatercept (ACE-536) | Celgene and Acceleron Pharma; ulticenter international sites |
| NCT03342404 | Phase 2; ongoing, not recruiting | 145 NTDT | Luspatercept (ACE-536) | Celgene and Acceleron Pharma; multicenter international sites |
| Trial identifier | Phase/status | Enrolled patients | Drug product | Sponsor/center |
|---|---|---|---|---|
| NCT01571635 | Phase 2; active, not recruiting | 16 TDT, 30 NTDT | Sotatercept (ACE-011) | Celgene; multicenter international sites |
| NCT01749540 | Phase 2; completed | 31 TDT, 33 NTDT | Luspatercept (ACE-536) | Acceleron Pharma; multicenter international sites |
| NCT02604433 | Phase 3; completed, not recruiting | 336 TDT | Luspatercept (ACE-536) | Celgene and Acceleron Pharma; ulticenter international sites |
| NCT03342404 | Phase 2; ongoing, not recruiting | 145 NTDT | Luspatercept (ACE-536) | Celgene and Acceleron Pharma; multicenter international sites |
From www.clinicaltrials.gov. In all studies, the route of administration was subcutaneous, and the target was ineffective erythropoiesis.
The BELIEVE trial is a randomized, double-blind, placebo-controlled phase 3 trial performed at 65 sites in 15 countries (Australia and countries across Europe, the Middle East, North Africa, North America, and Southeast Asia). Three hundred thirty-six patients aged ≥18 years who had confirmed β-thalassemia or Hb E/β-thalassemia and were regularly receiving transfusions (6-20 RBC units, with no transfusion-free period of >35 days within 24 weeks before randomization) were included in the trial. Patients were randomly assigned in a 2:1 ratio to receive luspatercept or placebo subcutaneously every 21 days for ≥48 weeks. The starting dose of luspatercept was 1.0 mg/kg of body weight with titration up to 1.25 mg/kg, according to the protocol. All the patients also received best supportive care, including RBC transfusions and iron chelation therapy, according to local guidelines. The primary end point was a ≥33% reduction in transfusion burden (with a reduction of ≥2 RBC units) during weeks 13 to 24, when compared with a 12-week period before randomization (baseline). Other efficacy end points included reductions of ≥33% or ≥50% in transfusion burden during any 12- and 24-week interval and results of iron studies.24 Of 224 patients, 48 (21.4%) in the luspatercept group achieved the primary end points compared with the patients in the placebo group (4.5%; odds ratio [OR], 5.79; 95% CI, 2.24-14.97; P < .001) Moreover, more patients had a ≥33% reduction in transfusion burden during any rolling 12-week interval (70.5% vs 29.5%; OR, 5.69; 95% CI, 3.46-9.35) or any 24-week interval (41.1% vs 2.7%; OR, 25.02; 95% CI, 7.76-80.71) with luspatercept vs placebo (Figure 2). Transfusion independence was achieved by 11% of the patients in the luspatercept group during any 8-week interval.24 Luspatercept showed a significant benefit over placebo across all prespecified subgroups: age, sex, origin, splenectomy, genotype, pretransfusion Hb, and iron level. Although benefit was observed in all subgroups, analyses of the genotype subgroups suggests that the magnitude of response to luspatercept is lower in patients with a β0/β0 genotype than in those with a non-β0/β0 genotype.25 The median time to the first response with luspatercept was within the first treatment cycle (12.0 or 24.5 days among the patients who had reductions in transfusion burden of ≥33% or ≥50%, respectively, during any 12-week interval). Most patients (80.4%) in the luspatercept group who had a reduction in transfusion burden of ≥33% from baseline during any 12-week interval had ≥2 distinct episodes of response, and 51.3% had ≥4 episodes of response. Similarly, 68.9% of the patients in the luspatercept group who had a ≥50% reduction in transfusion burden in any 12-week interval had ≥2 distinct responses, and 33.3% had ≥4 responses (Figure 3).24 The median longest duration of response with luspatercept was 104 days or 98 days among the patients who had reductions in the transfusion burden from baseline of at least 33% (158 patients) or at least 50% (90 patients), respectively, during any 12-week interval (Figure 4).24 Patients who completed the 48-week double-blind treatment period continued to receive luspatercept or placebo in a double-blind manner until all had completed the initial 48-week period. The trial group assignments were then unmasked, after which the patients randomized to placebo were eligible to cross over to receive luspatercept in an open-label phase. As of 1 July 2019, median treatment duration for patients in the luspatercept and placebo arms (before crossover) was 119.1 and 74.7 weeks, respectively. Of the patients initially randomized to the luspatercept arm, 68.2% were still receiving treatment at the end of 2 years. These patients continued to experience reductions in transfusion burden for 2 years.26
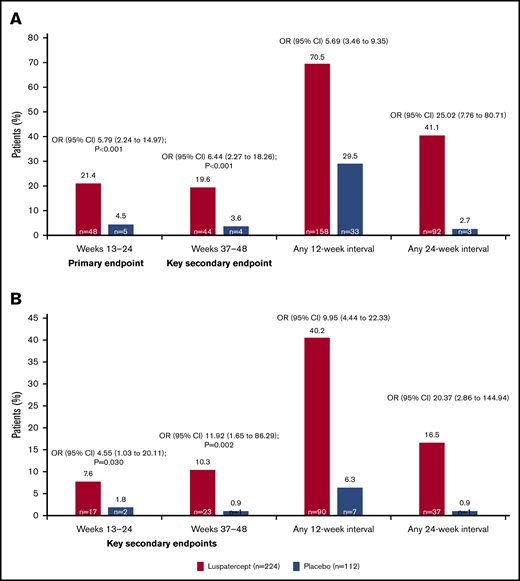
Percentage of patients who had a reduction in the transfusion burden of at least 33% or at least 50% from baseline. Reductions in the transfusion burden (defined as the total number of red-cell units transfused in a specified time interval) were assessed in the intention-to-treat population. (A) The percentages of patients who had a reduction in the transfusion burden of at least 33% from baseline during weeks 13 through 24 (primary end point), during weeks 37 through 48 (first key secondary end point), and during any 12-week or 24-week interval. (B) The percentages of patients who had a reduction in the transfusion burden of at least 50% from baseline during weeks 13 through 24 (second key secondary end point), during weeks 37 through 48 (third key secondary end point), and during any 12-week or 24-week interval. A reduction of at least 2 red-cell units over the fixed and nonfixed 12-week intervals was also required for those end points. To control for multiple comparisons, key secondary end points were evaluated in sequential order once the primary efficacy analysis had shown statistical significance. Reprinted with permission from Cappellini et al.24 Copyright © 2020 Massachusetts Medical Society.
Percentage of patients who had a reduction in the transfusion burden of at least 33% or at least 50% from baseline. Reductions in the transfusion burden (defined as the total number of red-cell units transfused in a specified time interval) were assessed in the intention-to-treat population. (A) The percentages of patients who had a reduction in the transfusion burden of at least 33% from baseline during weeks 13 through 24 (primary end point), during weeks 37 through 48 (first key secondary end point), and during any 12-week or 24-week interval. (B) The percentages of patients who had a reduction in the transfusion burden of at least 50% from baseline during weeks 13 through 24 (second key secondary end point), during weeks 37 through 48 (third key secondary end point), and during any 12-week or 24-week interval. A reduction of at least 2 red-cell units over the fixed and nonfixed 12-week intervals was also required for those end points. To control for multiple comparisons, key secondary end points were evaluated in sequential order once the primary efficacy analysis had shown statistical significance. Reprinted with permission from Cappellini et al.24 Copyright © 2020 Massachusetts Medical Society.
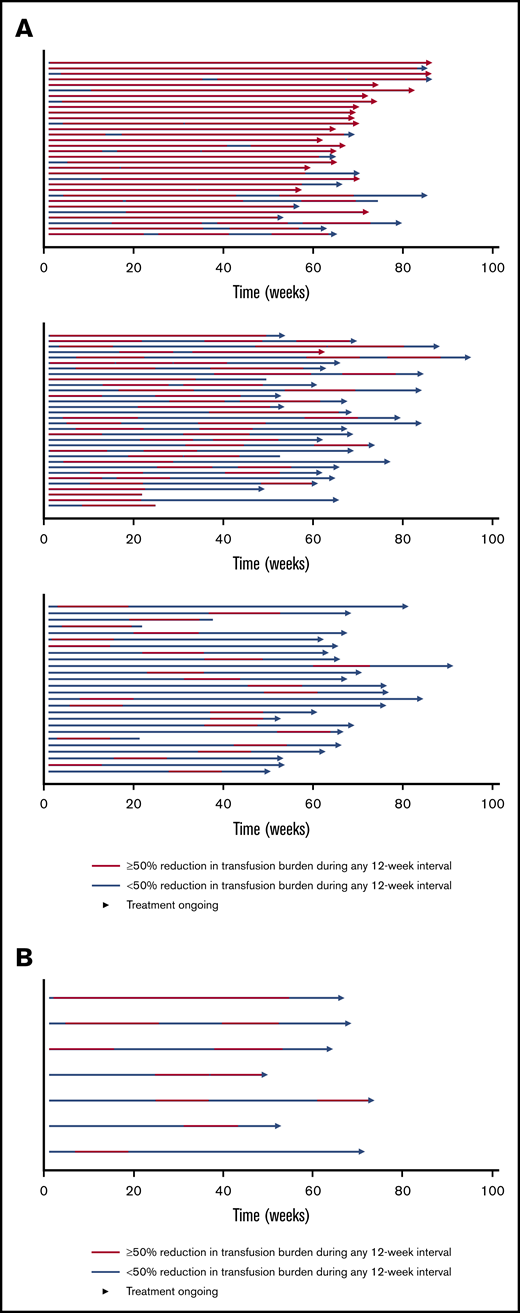
Swimmer plot of response periods for patients who had a reduction in the transfusion burden of at least 50% from baseline during any 12-week interval in the intention-to-treat population. Each row (swim lane) on the y-axis represents an individual patient in the luspatercept group (A) or the placebo group (B). A response period was defined as a continuous period in which a patient had a reduction in the transfusion burden of at least 50% from baseline during any 12-week interval. Different response periods may have overlapped. All patients received best supportive care in addition to luspatercept or placebo. Reprinted with permission from Cappellini et al.24 Copyright © 2020 Massachusetts Medical Society.
Swimmer plot of response periods for patients who had a reduction in the transfusion burden of at least 50% from baseline during any 12-week interval in the intention-to-treat population. Each row (swim lane) on the y-axis represents an individual patient in the luspatercept group (A) or the placebo group (B). A response period was defined as a continuous period in which a patient had a reduction in the transfusion burden of at least 50% from baseline during any 12-week interval. Different response periods may have overlapped. All patients received best supportive care in addition to luspatercept or placebo. Reprinted with permission from Cappellini et al.24 Copyright © 2020 Massachusetts Medical Society.
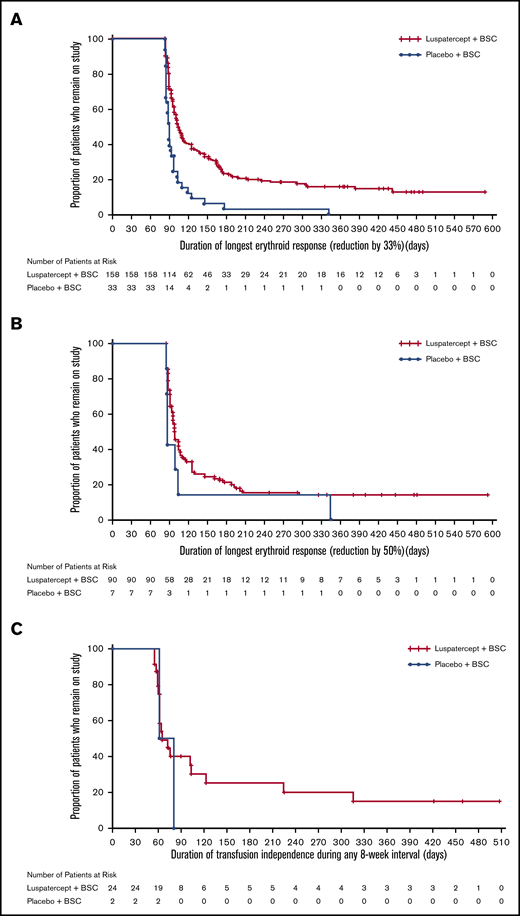
Kaplan-Meier plot of duration of response in responding patients achieving ≥33% transfusion reduction (any 12-week interval), ≥50% transfusion reduction (any 12-week interval), and transfusion independence (any ≥8-week interval). Only patients who achieved an erythroid response were included in the Kaplan–Meier analyses. Transfusion reduction was defined as ≥33% (A) or ≥50% (B) reduction in total RBC units from baseline (12 weeks prior to randomization) and ≥2 RBC units in any consecutive 12-week intervals throughout the study period. (C) Transfusion independence was defined as the absence of any transfusion during any consecutive 8-week intervals throughout the study period. BSC denotes best supportive care, RBC red blood cell. Reprinted with permission from Cappellini et al.24 Copyright © 2020 Massachusetts Medical Society.
Kaplan-Meier plot of duration of response in responding patients achieving ≥33% transfusion reduction (any 12-week interval), ≥50% transfusion reduction (any 12-week interval), and transfusion independence (any ≥8-week interval). Only patients who achieved an erythroid response were included in the Kaplan–Meier analyses. Transfusion reduction was defined as ≥33% (A) or ≥50% (B) reduction in total RBC units from baseline (12 weeks prior to randomization) and ≥2 RBC units in any consecutive 12-week intervals throughout the study period. (C) Transfusion independence was defined as the absence of any transfusion during any consecutive 8-week intervals throughout the study period. BSC denotes best supportive care, RBC red blood cell. Reprinted with permission from Cappellini et al.24 Copyright © 2020 Massachusetts Medical Society.
At week 48, a modest decrease of serum ferritin level (mean ±standard deviation change, −248 ± 800 μg/L) compared with baseline was observed in the luspatercept group. On the contrary, a mild increase from baseline in the placebo group was observed (mean change, 107 ± 526 μg/L). No clinically meaningful changes from baseline in liver iron concentration or myocardial iron deposition were observed during the assessment period. Longer follow-up is necessary to determine the impact of luspatercept on iron overload and use of iron chelation therapy.
Adverse events of transient bone pain, arthralgia, dizziness, hypertension, and hyperuricemia were more frequent with luspatercept than with the placebo. At least 1 serious adverse event was reported during the treatment period in 15.2% (95% CI, 10.8-20.6) of the patients in the luspatercept group and in 5.5% (95% CI, 2.0-11.6) of the patients in the placebo group.24 In 8 patients (3.6%) in the luspatercept group, clinically confirmed thromboembolic adverse events occurred during the treatment period (including 2 grade ≥3 events), and in 1 patient (0.9%) in the placebo group. All such events occurred in patients who had undergone splenectomy and had ≥1 other risk factor for thromboembolic disease, including a history of venous thrombosis or thrombocytosis at baseline.24 Although these thrombotic events mainly occurred in patients with known risk factors, monitoring patients for signs and symptoms of thrombotic events during therapy is recommended.
A 5-year, open-label extension phase is ongoing to provide long-term efficacy and safety data.
A phase 2 trial (BEYOND; www.clinicaltrials.gov; #NCT03342404) in adults with NTDT is ongoing and has completed enrollment. The primary end point is the proportion of patients with an increase in mean Hb concentration of ≥1 g/dL in the absence of RBC transfusion from weeks 13 to 24 vs baseline. A pediatric study (ACE-B-THAL-004; #NCT04143724) is ready to start enrollment but has been paused because of the coronavirus pandemic.
In summary, luspatercept is a first-in-class approved erythroid maturation agent, promoting late-stage RBC maturation and reducing the transfusion burden in TDT. On 8 November 2019, the FDA approved luspatercept (Reblozyl; Celgene Corp.) for treatment of anemia in adult patients with β-thalassemia who require regular RBC transfusions.5 The European Commission approved luspatercept on 25 June 2020, after a positive opinion, issued on 30 April 2020 by the Committee for Medicinal Products for Human Use of the European Medicines Agency, recommending approval of the drug.6,27 Although a clinical benefit of luspatercept was observed in all patient subgroups, the percentage of patients who had a response was greater in those with a non-β0/β0 genotype than in those with a β0/β0 genotype. This latter group remains a challenge in TDT management, and future directions should look into the combinatorial effects of multiple therapies. There are several questions that should be further evaluated with regard to luspatercept. The translated benefit of transfusion reduction into lower iron indices was mainly observed with serum ferritin levels, but changes in target organ concentrations were not clear. These changes should be further clarified with long-term observation considering the mechanisms of iron loading, given that unloading from the heart and liver can be slow. Careful analyses of these findings in the context of iron chelation received is needed. Last, real-world application will help inform how reduction in transfusion burden will be applied by physicians (reduction of number of units per transfusion visit, or delay in transfusion visits) and how it will affect patients’ quality of life.
Other indications and future directions
Luspatercept has also been approved recently by the FDA for the treatment of anemia that fails to respond to an erythropoiesis-stimulating agent and requires ≥2 RBC units over 8 weeks in adult patients with very low-to-intermediate–risk MDS with ring sideroblasts (MDS-RS) or with MDS/myeloproliferative neoplasm with RS and thrombocytosis (MDS/MPN-RS-T).28 In a phase 2, multicenter, dose-finding study (PACE-MDS; www.clinicaltrials.gov; #NCT01749514, extension study #NCT02268383) patients with low or intermediate 1-risk MDS or nonproliferative chronic myelomonocytic leukemia, received luspatercept subcutaneously once every 3 weeks at dose concentrations ranging from 0.125 to 1.75 mg/kg. An erythroid response (defined as a reduction in RBC transfusions of ≥4 units per 8 weeks in patients with a baseline transfusion burden of ≥4 units per 8 weeks or as an increase in Hb level of ≥1.5 g/dL over 8 weeks in patients with a baseline transfusion burden of <4 units per 8 weeks) was observed in 63% of luspatercept-treated patients, and 38% achieved transfusion independence for ≥8 weeks.29 Because the overall erythroid response rate was higher among patients with RS (≥15% RS) than other subtypes of MDS, the phase 3 trial enrolled patients with lower risk MDS-RS who had been receiving regular RBC transfusions and were refractory or unlikely to respond to erythropoiesis-stimulating agents.
The MEDALIST trial (www.clinicaltrials.gov; #NCT02631070) is a multicenter, randomized, double-blind, placebo-controlled trial that enrolled 229 patients randomly assigned in a 2:1 ratio to receive luspatercept or placebo, administered subcutaneously every 3 weeks for 24 weeks. Transfusion independence for ≥8 weeks was observed in 38% of the patients in the luspatercept group, compared with 13% of patients in the placebo group (P < .001). During the first 24 weeks, 28% of patients in the luspatercept group had transfusion independence for ≥12 weeks, compared with 8% of patients in the placebo group, and the corresponding values during weeks 1 through 48 were 33% and 12%, respectively (P < .001). Also, a higher percentage of patients in the luspatercept group than in the placebo group had transfusion independence for ≥16 weeks or longer.30,31
Considering the potential mechanism of action, the use of luspatercept could be explored in other types of anemia that are associated with ineffective erythropoiesis or dyserythropoiesis.
Original data are available by e-mail request to the corresponding author.
Acknowledgments
The authors thank the patients and families who participated in the luspatercept clinical trial and the investigators who collaborated in the trials, Daniela Canali for drawing Figure 1, and Khaled Musallam for invaluable support in reviewing the manuscript.
Authorship
Contribution: M.D.C. and A.T.T. prepared and drafted the manuscript, made critical revisions, and approved the final version of the manuscript.
Conflict-of-interest disclosure: M.D.C. has served on advisory boards for BMS/Celgene, CRISPR, Novartis, Novonordisk, Sanofi/Genzyme, Silence, and Vifor. A.T.T. has provided consultancy for Abfero Pharmaceuticals Inc, Ionis Pharmaceuticals, La Jolla Pharmaceuticals, Novartis, and Protagonist Therapeutics; has received research funding from Bristol Myers Squibb, La Jolla Pharmaceuticals, Novartis, and Protagonist Therapeutics; and has received honoraria from Novartis.
Correspondence: Maria Domenica Cappellini, University of Milan, Via F. Sforza 35, 20122 Milan Italy; e-mail: maria.cappellini@unimi.it.

