

Key Points
Some patients with UCD present with an MCD-like inflammatory state, including constitutional symptoms and laboratory abnormalities.
When surgical excision is possible, UCD with MCD-like inflammatory state responds well.

Abstract
Unicentric Castleman disease (UCD) is a rare lymphoproliferative disorder presenting as a single nodal mass with characteristic histopathology. Patients with UCD are typically asymptomatic with normal laboratory markers, whereas patients with multicentric Castleman disease (MCD) demonstrate multicentric lymphadenopathy and cytokine storm–induced systemic inflammatory symptoms. This retrospective analysis of 116 UCD cases identified 19 (16.4%) cases with an MCD-like inflammatory state (UCD-MIS). We compared treatments and outcomes between cases of UCD-MIS and UCD–non-MIS to evaluate the role of surgery and illuminate biological behavior of UCD-MIS. There were differences in the distribution of histopathological subtypes (plasmacytic histopathology was more frequently seen, 52.6% vs 13.4%; P < .001) between the 2 groups. However, both groups demonstrated good responses to surgical treatment, suggesting that UCD-MIS in some patients still shared common biological behavior with UCD in other patients. Sixteen (94.2%) patients with UCD-MIS underwent complete surgical excision alone, and the systemic inflammation resolved completely in all of them. This high response rate suggests surgical treatment as a potential cure for this unique subset of patients. After a median follow-up duration of 64 months (range, 2-239 months), neither lymphadenopathy nor the inflammatory state recurred. However, inflammation may progress in patients with irresectable disease, and treatment options other than surgery should be considered in these patients.
Introduction
Castleman disease (CD) is a group of rare lymphoproliferative disorders featuring a common spectrum of histopathology, from regressed germinal centers and prominent vascularization (hyaline vascular [HV]) to hyperplastic germinal centers with prominent plasmacytosis (plasmacytic [PC]).1 It comprises unicentric CD (UCD), in which only a single lymph node region is enlarged, and multicentric CD (MCD) with multiple regions of enlarged lymph nodes, systemic inflammation, and life-threatening organ dysfunction related to a cytokine storm.
The etiology of UCD is currently unknown. Recent studies have identified somatic mutations in lymph node tissue from patients with UCD, specifically PDGFRb mutations in lymph node stromal cells that may give rise to UCD.2 In contrast to idiopathic MCD (iMCD), which is characterized by systematic inflammatory symptoms, patients with UCD are usually asymptomatic or present with only local compressive symptoms, and laboratory test results are usually normal.3 Complete surgical resection carries a 10-year disease-free survival of 90% to 95%4,5 and is considered the gold standard of treatments for UCD. Other potential effective therapeutic approaches, including radiation, embolization, and neoadjuvant chemotherapy or immunotherapy, may be considered when the lymph node is irresectable.1 The disease usually demonstrates a benign course, with a reported 5-year overall survival (OS) of 92% to 99%.4-9
Meanwhile, we have observed in clinical practice that a portion of patients with UCD present with severe constitutional symptoms accompanied by an inflammatory state resembling iMCD in clinical presentations and laboratory test results. Oksenhendler et al also reported that 9 of 16 patients with UCD with the PC variant were symptomatic and seemed to have MCD.7 The diagnosis and treatment of these patients are challenges for clinicians to decide whether their disease still belongs to the UCD subtype, in which case, they could be readily cured by surgical treatment. Case reports have shown that systematic symptoms can be eliminated by complete resection of the enlarged lymph nodes.10-12 However, because of the rarity of cases, a comprehensive study of this subgroup, their response to surgery, and long-term outcome is currently unavailable. In fact, it is unknown whether surgical excision should still serve as the first-line treatment option in these patients, or they should receive systemic treatments typically used for iMCD.
We performed a retrospective study to characterize the clinical features, treatments, and outcomes of patients with UCD with an MCD-like inflammatory state (UCD-MIS), to compare them with other patients with UCD (UCD–non-MIS) and illuminate the biological behavior of the disease in these cases and delineate the value of surgical treatment.
Materials and methods
We identified 127 patients in the hospital information system of Peking Union Medical College Hospital who had a UCD diagnosis and were treated from 1 January 2000 through 31 December 2019. The diagnosis of UCD was reconfirmed by reexamination of the pathological specimens and imaging reports according to the algorithm adapted from Fajgenbaum et al.13 Patients with classic CD histopathology and 1 (unicentric) or >1 (oligocentric) enlarged lymph node found in a single region (on one side of the neck and in the mediastinum, abdomen, and retroperitoneum, among other sites) were included. Two patients were excluded after a reexamination of pathological specimens that were not consistent with classic CD histopathological findings. Computed tomographic (CT) scan or magnetic resonance imaging including neck, chest, abdomen, and pelvis or fluorodeoxyglucose positron emission tomography/CT reports were retrieved from the database to confirm the centricity of the disease (unicentric or oligocentric). Because malignancies and chronic infection may impact the inflammatory state, 1 patient with follicular dendritic cell sarcoma and one with tuberculosis were excluded. Because of the unique disease presentation and treatment of paraneoplastic pemphigus and bronchiolitis obliterans, patients with disease complicated with those conditions were excluded, resulting in a total of 116 patients eligible for this study (Figure 1).
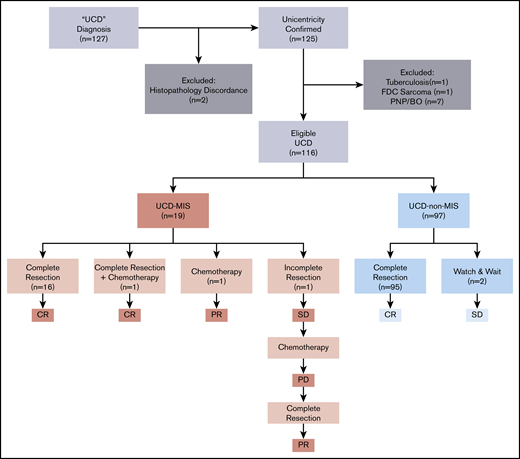
We identified UCD-MIS based on clinical and laboratory criteria borrowed from the diagnostic criteria of iMCD.13 Clinical criteria include constitutional symptoms, hepatosplenomegaly, edema or pleural effusions, eruptive cherry hemangiomatosis or violaceous papules, and lymphocytic interstitial pneumonitis. Laboratory criteria included elevated c-reactive protein (CRP), anemia, thrombocytopenia or thrombocytosis, hypoalbuminemia, renal dysfunction, and polyclonal hypergammaglobulinemia. Patients meeting at least 2 criteria with at least 1 laboratory abnormality, which mirrors the requirement for a diagnosis of iMCD, were included in the UCD-MIS group.
Demographic, clinical, laboratory and treatment-related data were extracted from patients’ medical records, including age, sex, symptoms at presentation, physical examination, imaging, laboratory test, histopathology, treatment strategies, and response. Follow-up information was obtained from outpatient clinics, phone contacts, and records of visits in hospital databases.
To evaluate the treatment response of UCD-MIS in symptoms and laboratory abnormalities, we referred to the response criteria of iMCD defined by the Castleman Disease Collaborative Network.14 Complete remission was defined as normalization of laboratory test results and inflammation-related symptoms, including fatigue, anorexia, fever, and weight loss. Partial remission was defined as improvement in all 4 symptom categories, >50% improvement in laboratory results, and >50% reduction of the mass. Progressive disease was defined as >25% deterioration in laboratory test results, aggravation of any of 4 inflammation-related symptoms, >25% increase or reoccurrence of the enlarged lymph node, or development of an MIS (in patients with no inflammation). Stable disease was defined as no partial or complete remission or progressive disease. OS time was the period from diagnosis to date of death or the last follow-up. Patients were followed up until 31 December 2019.
We summarized patient characteristics by descriptive analysis. UCD-MIS was compared with UCD–non-MIS using the χ2, Fisher exact, Student t, and Mann-Whitney tests. OS was assessed by Kaplan-Meier analysis, and comparison between the 2 groups was made with the log-rank test. P < .05 was considered statistically significant. All statistical analyses were performed with SPSS version 25.0 for Windows (IBM, Armonk, NY).
The study was performed with prior approval of the Institutional Review Board and the Ethics Committee of the Peking Union Medical College Hospital.
Results
Baseline characteristics
A total of 116 patients with histopathologically and radiologically confirmed UCD were enrolled in the study, consisting of 45 males and 71 females, with a median age of 39 years (range, 13-79 years) at the time of diagnosis. Nineteen patients (16.4%) were identified as having UCD-MIS. The other 97 patients (93.6%) who did not meet the inclusion criteria formed the UCD–non-MIS group. There were no significant differences in age and sex distribution between the 2 groups (Table 1).
Demographic and disease characteristics of patients with UCD, with and without MIS
| MIS (n = 19) | Non-MIS (n = 97) | P | |
|---|---|---|---|
| Age, mean ± SD, y | 37.8 ± 14.9 | 39.9 ± 14.4 | .567 |
| Sex, n (%) | .532 | ||
| Male | 7 (36.8) | 38 (39.2) | — |
| Female | 12 (63.2) | 59 (60.8) | — |
| Histopathology, n (%) | <.001 | ||
| HV | 6 (31.6) | 77 (79.4) | <.001 |
| Mix | 3 (15.8) | 7 (7.2) | .21 |
| PC | 10 (52.6) | 13 (13.4) | <.001 |
| Oligocentric disease, n (%) | 3 (15.8) | 2 (2.1) | .031 |
| Inoperable, n (%) | 2 (10.5) | 2 (2.1) | .125 |
| MIS (n = 19) | Non-MIS (n = 97) | P | |
|---|---|---|---|
| Age, mean ± SD, y | 37.8 ± 14.9 | 39.9 ± 14.4 | .567 |
| Sex, n (%) | .532 | ||
| Male | 7 (36.8) | 38 (39.2) | — |
| Female | 12 (63.2) | 59 (60.8) | — |
| Histopathology, n (%) | <.001 | ||
| HV | 6 (31.6) | 77 (79.4) | <.001 |
| Mix | 3 (15.8) | 7 (7.2) | .21 |
| PC | 10 (52.6) | 13 (13.4) | <.001 |
| Oligocentric disease, n (%) | 3 (15.8) | 2 (2.1) | .031 |
| Inoperable, n (%) | 2 (10.5) | 2 (2.1) | .125 |
Significant P values are in bold.
At presentation, 63.2% of the patients with UCD-MIS reported constitutional symptoms, 52.6% had hepatomegaly and/or splenomegaly and 26.3% had edema or pleural effusions. Of all the patients with UCD-MIS, anemia was found in 72.2%, abnormal platelet count in 50.0% (33.3% elevated and 16.7% decreased), albumin decrease in 22.2%, renal function impairment in 17.6%, erythrocyte sedimentation rate (ESR) elevation in 66.7%, CRP elevation in 77.9%, immunoglobulin G (IgG) increase in 77.9%, and interleukin-6 (IL-6) elevation in 50.0% (Table 2).
Clinical characteristics of patients with UCD with and without MIS
| MIS | Non-MIS | |||
|---|---|---|---|---|
| Patients, n | n (%) | Patients, n | n (%) | |
| Constitutional symptoms* | 19 | 12 (63.2) | 97 | 8 (8.2) |
| Hepatomegaly and/or splenomegaly | 19 | 10 (52.6) | 97 | 2 (2.1) |
| Fluid accumulation† | 19 | 5 (26.3) | 97 | 1 (1.0) |
| Anemia‡ | 18 | 13 (72.2) | 87 | 4 (4.6) |
| Abnormal PLT§ | 18 | 9 (50.0) | 87 | 4 (4.6) |
| Thrombocytosis | — | 6 (33.3) | — | 3 (3.4) |
| Thrombocytopenia | — | 3 (16.7) | — | 1 (1.1) |
| Albumin <35, g/L | 9 | 2 (22.2) | 30 | 0 (0.0) |
| eGFR <60.00, mL/min per 1.73 m2 | 17 | 3 (17.6) | 83 | 0 (0.0) |
| ESR >15, mm/h | 9 | 6 (66.7) | 22 | 5 (22.7) |
| hsCRP >3.00, mg/L | 9 | 7 (77.8) | 22 | 4 (18.2) |
| IgG >17.00, g/L | 12 | 7 (77.8) | 23 | 2 (8.7) |
| IL-6 >5.9, pg/mL | 10 | 5 (50.0) | 20 | 2 (10.0) |
| MIS | Non-MIS | |||
|---|---|---|---|---|
| Patients, n | n (%) | Patients, n | n (%) | |
| Constitutional symptoms* | 19 | 12 (63.2) | 97 | 8 (8.2) |
| Hepatomegaly and/or splenomegaly | 19 | 10 (52.6) | 97 | 2 (2.1) |
| Fluid accumulation† | 19 | 5 (26.3) | 97 | 1 (1.0) |
| Anemia‡ | 18 | 13 (72.2) | 87 | 4 (4.6) |
| Abnormal PLT§ | 18 | 9 (50.0) | 87 | 4 (4.6) |
| Thrombocytosis | — | 6 (33.3) | — | 3 (3.4) |
| Thrombocytopenia | — | 3 (16.7) | — | 1 (1.1) |
| Albumin <35, g/L | 9 | 2 (22.2) | 30 | 0 (0.0) |
| eGFR <60.00, mL/min per 1.73 m2 | 17 | 3 (17.6) | 83 | 0 (0.0) |
| ESR >15, mm/h | 9 | 6 (66.7) | 22 | 5 (22.7) |
| hsCRP >3.00, mg/L | 9 | 7 (77.8) | 22 | 4 (18.2) |
| IgG >17.00, g/L | 12 | 7 (77.8) | 23 | 2 (8.7) |
| IL-6 >5.9, pg/mL | 10 | 5 (50.0) | 20 | 2 (10.0) |
Significant P values are in bold.
eGFR, estimated glomerular filtration rate; hsCRP, high-sensitivity C-reactive protein; PLT, platelet count.
Night sweats, fever (>39°C), weight loss, or fatigue (≥2 Common Terminology Criteria for Adverse Events lymphoma score for B-symptoms).
Edema, anasarca, ascites, or pleural effusion.
Hemoglobin: <12 g/dL in males, <11 g/dL in females.
Thrombocytopenia (PLT <100 k/μL) or thrombocytosis (PLT >350 k/μL).
A significant difference was also observed between the histopathological subtypes in the 2 groups of patients. The HV subtype dominated in UCD–non-MIS (79.4% vs 31.6%; P < .001), whereas the PC subtype was more common in UCD-MIS (52.6% vs 13.4%; P < .001; Table 1).
In UCD-MIS, the enlarged lymph node was most commonly present in the mediastinal (38.9%), retroperitoneal (33.3%), and abdominal (16.7%) regions (Figure 2A). In UCD–non-MIS, the enlarged lymph node occurred most frequently in the mediastinal (29.9%), retroperitoneal (23.9%), cervical (19.0%), and abdominal (17.9%) regions (Figure 2B). There was more oligocentric disease in the UCD-MIS group (15.9% vs 2.1%, P = .031). Meanwhile, the difference in the percentage of patients with inoperable disease in the 2 groups was not significant (10.5% vs 2.1%; P = .125; Table 1).

Involved lymph node regions. Patients with UCD with MIS (A) and without MIS (B).
Involved lymph node regions. Patients with UCD with MIS (A) and without MIS (B).
Treatments and outcomes
Complete surgical excision alone was the first-line treatment in 95 of 97 (97.9%) cases of UCD-non–MIS, with no recurrence of lymphadenopathy. The disease was inoperable in 2 asymptomatic patients, to whom a watch-and-wait strategy was applied, and no disease progression was seen (Figure 1).
Meanwhile, 16 of 19 UCD-MIS (94.2%) underwent complete surgical excision (Figure 1). No recurrence of lymphadenopathy was seen after surgery, and the inflammation resolved completely in all 16 patients. A whole-body contrast CT scan including neck, chest, abdomen, and pelvis was performed in these patients, and no newly developed mass was discovered. Hepatomegaly and/or splenomegaly improved but did not normalize in 5 of 8 patients after surgery. One patient with splenomegaly underwent splenectomy before lymphadenectomy. No significant difference was seen in the other 2 patients with organomegaly after surgery. One patient with severe constitutional symptoms (grade 3 fatigue, grade 2 anorexia, and grade 2 weight loss, according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0) was prescribed a thalidomide-cyclophosphamide-prednisone (TCP) regimen 3 weeks after surgery and achieved a complete remission. In 2 patients (10.5%), complete surgical resection was impossible at the time of diagnosis. One was treated with systemic chemotherapy alone (TCP regimen). The inflammation resolved, with >50% reduction of the mass and no significant improvement in splenomegaly, and the patient reached partial remission. In the other patient, tumor reduction surgery was performed at first without any significant improvement in inflammation-related symptoms and biomarkers, and the TCP regimen was initiated 6 months after surgery. The inflammation partially resolved but then progressed 3 months after the start of chemotherapy. The patient was reevaluated for surgery and underwent complete resection of the mass and several enlarged lymph nodes in the same region (retroperitoneal). Partial remission was reached at last follow-up, with laboratory test results and the hepatosplenomegaly improved, but not normalized.
Survival analysis
The median follow-up duration was 64 months (range, 2-239). The difference between the median follow-up time of the 2 groups was not statistically significant (69.4 vs 49.1 months; P = .705). The OS between the 2 groups was not significantly different (P = .691; HR, 3.22; 95% CI, 0.012-943).
Discussion
It has been reported that some patients with UCD, despite the unicentricity of the disease, present with MCD-like constitutional symptoms and abnormal laboratory results, but this unique subset of patients has not been well delineated, and their clinical features and treatment response required further illumination. Thus, our retrospective series of 19 patients with inflammatory UCD is one of the first endeavors that we are aware of to provide in-depth information on this potential subgroup of UCD. Our study showed that UCD-MIS responded well to surgical excision when possible, which cured all lymphadenopathies and symptoms and abnormal laboratory results. These results support those in previous studies that demonstrated the role of surgical resection in UCD, suggesting that the pathology of UCD was localized to the lymph node tissue regardless of inflammatory state. Despite some clinical manifestations that mimic MCD, UCD should still be treated with surgical excision whenever possible.
The good response of UCD-MIS to surgical excision of the solitary enlarged lymph node further supports the difference in etiology between UCD and iMCD. In patients with iMCD, the enlarged lymph nodes and histopathological features are considered to be reactive changes in response to the cytokine storm, with elevated IL-6 and other circulating factors,15 and treatments for iMCD are directed at controlling hyperactivated immune cells and containing the cytokine storm. The treatment response to surgical resection in our UCD cohort shows that the systemic inflammation that can be seen in UCD is related to the cell population present in the enlarged lymph node. The frequency of oligocentric disease also supports this idea. This finding is consistent with previous reports of somatic mutations in lymph node tissue from patients with UCD.2 Further investigations into the pathogenesis of UCD, including genetic predispositions, signaling pathways, and cell type dysregulations, are needed, to understand the cause of different biological behavior but similar histopathological and, in some cases, clinical presentations of UCD and iMCD.
In the 2 inoperable cases of UCD-MIS, 1 patient responded fairly well to systemic chemotherapy. The other, who experienced disease progression while receiving chemotherapy, was able to undergo radical surgery after reevaluation. Therefore, clinicians treating UCD-MIS should consider systemic approaches typically used for iMCD when complete surgical resection is considered impossible, either as a sole treatment or as a neoadjuvant therapy before a radical excision. The TCP regimen used in these patients is an effective treatment option for iMCD,16 but its effectiveness in inoperable UCD-MIS needs further validation. The use of radiotherapy has also been reported, with a different response rate,5,17,18 but it was not used in our cohort because of concern for acute and long-term toxicity.
Our study also revealed that PC histopathology was more common in UCD-MIS than was HV histopathology. Previous studies have suggested that the PC histopathological subtype of UCD is more likely to be associated with the systemic symptoms and abnormal laboratory findings19 and an increase in serum IL-6, similar to iMCD. Boutboul et al, in a study of 71 cases of UCD, reported that there was a higher percentage of anemia, hypoalbuminemia, and CRP elevation in patients with PC or mixed histopathology; half (8 of 16) of these patients were symptomatic, and their disease was similar to iMCD.5 Further research is needed to uncover the mechanisms through which the inflammatory state occurs in patients demonstrating PC histopathology with treatment implications.
Nearly half of the patients had hepatomegaly and/or splenomegaly. They were more likely to experience anemia, abnormal platelet count, and elevated CRP, but the trend was not seen in ESR, IL-6, or IgG (results not shown). Because the organ enlargement improved in most patients after treatment, it may be true that it was secondary to the inflammatory state. However, the enlargement may not be completely reversible; therefore, it was not normalized in any of the patients, although their inflammatory state was completely or partially cured.
Patients with oligocentric disease were included in this study, and the smaller lymph nodes in the region of the bulk of the disease were considered “satellite” lymph nodes. Oksenhendler et al reported in 2018 that the clinical and biological characteristics of patients with several affected lymph nodes in a single site are very similar to those of patients with single lymph node involvement.7 After the bulk of the disease is surgically resected, the remaining smaller lymph nodes recess gradually.20 Although, in our patient cohort, those with oligocentric disease were more frequently seen in the UCD-MIS group, their good response to surgery suggested that they may be more accurately classified as UCD, not iMCD.
In clinical settings, the diagnosis of UCD may be more challenging when MIS is present, if clinicians are not aware of these cases. Therefore, thorough physical examination of peripheral lymph nodes and whole-body CT scan or positron emission tomography/CT scan, including neck, chest, abdomen, and pelvis, should be performed in this subset of patients to confirm the unicentricity of disease.1,4 If local disease is confirmed, this study supports surgery as the first-line therapy, regardless of the clinical and abnormal laboratory results that may be present.
There is currently no standardized response criteria for UCD, especially for those with MIS. In this study, we cited the response criteria of iMCD defined by the Castleman Disease Collaborative Network and made adaptations for patients with UCD-MIS to evaluate their treatment response. This evaluation criteria would also be suitable for patients with UCD-MIS in clinical settings.
The current study has a few limitations. First, because of the retrospective nature of the study, some laboratory data were not tested and thus were not available in some asymptomatic patients. Second, when patients presented with more severe symptoms, physicians may have selected more intensive treatments, such as chemotherapy together with surgical excision, thereby possibly achieving better treatment responses. The heterogeneity of treatment based on clinical characteristics may have biased the results of this study. Third, because our hospital is a referral center for rare diseases, the patient population in our cohort may not be representative of the true UCD population, and results from our cohort may overestimate the proportion of patients with MIS. Last, there may be differences in UCD presentations and disease course in different regions of the world.
In summary, UCD-MIS is not uncommon. Although patients demonstrate MCD-like inflammatory symptoms and more frequently have PC histopathology, their disease is still curable with complete surgical excision. This important clinical observation also has important implications with regard to understanding disease biology, as our study suggests that the pathological cell population is confined to lymph node tissue in these cases, despite the presence of systemic symptoms. In cases of irresectable disease, other treatment options such as chemotherapy should be considered in patients with an inflammatory state.
For the original data, please contact pumczhanglu@126.com.
Acknowledgments
The authors thank Shu-huai Zhang for assistance in preparing the graphics.
This work was supported by institutional research funding provided by the National Natural Science Foundation of China (grants 81974011 [J.L.] and 81900202 [L.Z.]), Fundamental Research Funds for the Central Universities (grant 3332018036) (L.Z.), Beijing Natural Science Foundation (grant 7182128) (J.L.), the Chinese Academy of Medical Science Innovation Fund for Medical Sciences (grant 2016-12M-1-002) (J.L.), and National Key Research and Development Program of China (grant 2016YFC0901503) (J.L.).
Authorship
Contribution: J.F., X.-x.C., D.-b.Z., L.Z., and J.L. recruited the patients; L.Z. and J.L. designed the study; M.-y.Z., M.-n.J., and J.C. collected the data; M.-y.Z. performed the analysis; M.-y.Z., L.Z., J.L., and D.C.F. interpreted the data and wrote the manuscript; and all authors had access to primary clinical trial data and gave final approval to submit for publication.
Conflict-of-interest disclosure: D.C.F. has received research funding from Janssen Pharmaceuticals and EUSA Pharma. The remaining authors declare no competing financial interests.
Correspondence: Lu Zhang, Department of Hematology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences–Peking Union Medical College, No. 1 Shuaifuyuan, Dongcheng District, Beijing 100730, China; e-mail: pumczhanglu@126.com; and Jian Li, Department of Hematology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences–Peking Union Medical College, No. 1 Shuaifuyuan, Dongcheng District, Beijing 100730, China; e-mail: lijian@pumch.cn.

