

Key Points
Regimen of ATRA + ATO + GO in high-risk APL is associated with prolonged EFS and high rate of early molecular response.
Regimen of ATRA + ATO + GO was safe and associated with a low rate of early death in high-risk APL.

Abstract
High-risk acute promyelocytic leukemia (APL) remains a therapeutic challenge, with higher associated rates of early mortality and relapse than standard-risk APL. All-trans retinoic acid (ATRA) plus arsenic trioxide (ATO) is a well-established treatment for patients with standard-risk APL, but it is not well defined for those with high-risk APL. In a prior study of patients with high-risk APL, the addition of gemtuzumab ozogamicin (GO) to ATO plus ATRA suggested benefit. The SWOG Cancer Research Network conducted a phase 2 study to confirm the efficacy and safety of the combination of ATRA plus ATO plus GO in treating high-risk APL patients. The primary end points were 3-year event-free survival (EFS) and early (6-week) death rates associated with this combination. Seventy patients were treated. With a median follow-up of 3.4 years, the 3-year EFS and overall survival estimates were 78% (95% confidence interval [CI], 67%-86%) and 86% (95% CI, 75%-92%), respectively. Overall, 86% of patients achieved complete response. The 6-week mortality rate was 11%. The most common treatment-emergent toxicities during the induction phase included febrile neutropenia, aspartate aminotransferase/alanine aminotransferase elevation, hyperglycemia, hypoxia, headache, and prolonged QT interval corrected for heart rate. Retinoic acid syndrome occurred in 9% of patients. Approximately 37% of patients did not complete all planned courses of postremission therapy. The combination of ATRA plus ATO plus GO in high-risk APL patients was effective and generally well tolerated, suggesting an opportunity to offer a chemotherapy-free induction platform for patients with this disease. This trial was registered at www.clinicaltrials.gov as #NCT00551460.
Introduction
Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia that is typically characterized by a balanced translocation between chromosomes 15 and 17 [t(15;17)(q22;q21)], a heightened risk of early hemorrhagic complications, and a generally favorable prognosis. Historically, high-risk APL (defined by a white blood cell count ≥10 × 103/μL) has been associated with increased risks of early hemorrhagic death and relapse.1,2
Although arsenic trioxide (ATO)–based regimens have clearly improved outcomes for patients with standard-risk APL,3,4 their effects on patients with high-risk APL are less clear. Recent trials from both the North American Intergroup Study and the Medical Research Council have confirmed the benefit of ATO as frontline therapy for patients with APL and have reported similar outcomes among smaller high-risk subgroups.5,6
Gemtuzumab ozogamicin (GO) is a humanized anti-CD33 antibody-drug conjugate that was initially approved for relapsed acute myeloid leukemia patients >15 years ago; after a temporary removal from the US commercial market in 2010, GO was reapproved in 2017. For patients with APL, GO is an attractive therapeutic strategy, given the universally high expression of CD33 in this disease. In an earlier pilot study of high-risk APL patients, GO was combined with all-trans retinoic acid (ATRA) plus ATO, resulting in acceptable safety and efficacy.7 To further identify the benefit of GO in combination with ATRA plus ATO during induction therapy, the SWOG Cancer Research Network conducted a clinical trial of this combination in patients with high-risk APL.
Patients and methods
Eligibility
All patients provided written informed consent to participate in this study. The trial was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines as defined by the International Conference on Harmonisation. The study protocol, amendments, and patient consent forms were approved by the institutional review board and/or independent ethics committee at each study site before the start of the trial. Patients age ≥18 years with newly diagnosed high-risk APL (PML-RARα+ by reverse transcriptase polymerase chain reaction [PCR]), defined by a white blood cell count ≥10 × 103/μL, were eligible. Additional guidelines for exclusion included other serious illnesses with a prognosis of <2 years, psychiatric conditions preventing treatment compliance or informed consent, severe or uncontrolled cardiovascular or pulmonary disease, abnormalities in baseline hepatic or renal function considered as serious obstacles to safe tolerance of therapy, and known pregnancy.
Treatment plan
The treatment schema is shown in Figure 1. Induction therapy consisted of ATRA plus ATO plus GO. After achieving CR, patients received 2 cycles of consolidation therapy with ATO, followed by 2 cycles of consolidation therapy with daunorubicin plus ATRA and then 2 cycles of consolidation therapy with GO. After completion of consolidation therapy, maintenance therapy with ATRA, 6-mercaptopurine, and methotrexate was administered for up to 1 year.
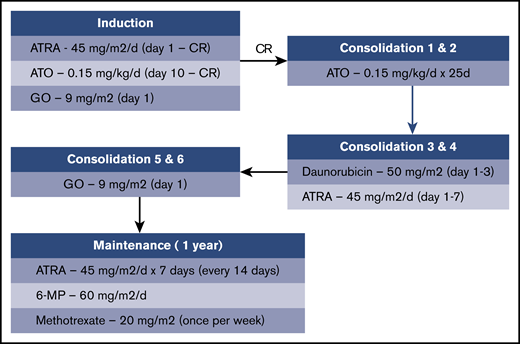
Supportive care guidelines included the use of antiinfective therapy per institutional standards, at least twice weekly monitoring of coagulation studies during induction until normalization and as clinically indicated, blood product support to maintain platelet count between 30 × 103/μL and 50 × 103/μL during the initial phases of therapy, cryoprecipitate to maintain a fibrinogen level of ≥100 mg/dL, and correction of electrolyte abnormalities to within normal limits before and during treatment with ATO. Growth factors could be administered at the discretion of the treating physician. Hydroxyurea administration was permitted up to 24 hours before the initiation of GO to control leukocytosis. Guidelines for the management of ATRA syndrome included the temporary discontinuation of ATRA and administration of dexamethasone for 3 days.
Patients who achieved CR after induction therapy were allowed to move on to consolidation therapy. During consolidation therapy, patients who maintained CR status after each even-numbered cycle of consolidation therapy could continue to the next phase of consolidation therapy and, ultimately, maintenance therapy. Molecular CR status was not required for entry into any phase of treatment.
Definitions and study end points
The dual primary end points of the study were the rate of event-free survival (EFS) at 3 years and the rate of mortality at 6 weeks. Secondary end points included the rate of toxicity of the regimen and the molecular response rate. CR was defined according to the revised criteria by Cheson et al.8 Molecular response was defined as the achievement of CR with the disappearance of the PML-RARα transcript detected via locally performed reverse transcriptase PCR assay. Response assessments were performed before treatment, after induction therapy (on or before day 42), before consolidation therapy cycle 3, and before maintenance therapy.
Statistical analyses
The primary objective of this study was to assess the 3-year EFS and 6-week mortality rates of patients treated with the described regimen. The joint null hypothesis was that the 3-year EFS rate was at most 50% and that the 6-week mortality rate was at least 30%. Three-year EFS was defined as the achievement of CR without relapse or death before 3 years after registration. Three patients who achieved CR withdrew consent without report of relapse and before completing 3 years of follow-up; these patients were not evaluable for the binary EFS end points. Therefore, for the primary analysis, the Kaplan-Meier method was used to produce unbiased estimates of 3-year EFS. To conduct the Kaplan-Meier analyses, EFS was measured from the date of study registration to the first date off treatment without a documented CR, the date of relapse from CR, or the date of death. Patients who were last known to be alive and relapse free were censored at the date of last contact. A sensitivity analysis was performed using the binary EFS end points and an exact binomial test. This analysis found that the 3 patients who withdrew consent did not achieve 3-year EFS. The 6-week mortality rate was analyzed using an exact binomial test.
Overall survival (OS) was measured from the date of study registration to the date of death resulting from any cause, with patients last known to be alive censored at the date of last contact. Relapse-free survival (RFS) was measured from the date of CR to the date of death or relapse, with patients last known to be alive and relapse free censored at the date of last contact. Landmark analyses of OS and RFS were performed among patients who achieved CR. To perform these analyses, OS and RFS times were landmarked at the maximum number of days observed from registration until completion of consolidation therapy (413 days after registration). OS and RFS were then compared using log-rank tests across 2 groups: patients who completed and patients who did not complete consolidation therapy. The cumulative incidence of relapse (CIR) was determined as the estimated probability of relapse up to a given time, considering death as a competing risk. Cox proportional hazards regression analyses were used to investigate the EFS rates of patients whose molecular response data were available within 8 weeks of diagnosis. A permutation test was used to calculate a P value between patients with and without a PCR response before 8 weeks.
Statistical analyses were performed using SAS (version 9.4; SAS Institute, Cary, NC) and R software (version 3.4.3). The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Efficacy
Between 2008 and 2013, 70 registered patients began protocol treatment. Patient demographics are shown in Table 1. Median age was 46.5 years (range, 19.2-86.4 years); 53% of patients were women. Of the 78 patients who were initially registered to the study, 70 were eligible to receive therapy. The study flow diagram (Figure 2) outlines the specific phases of treatment and reasons for discontinuation.
Baseline patient characteristics in S0535 (N = 70)
| Characteristic | n | % |
|---|---|---|
| Age, y | ||
| Median | 46.5 | |
| Range | 19.1-86.3 | |
| Baseline WBC count, × 103/μL | ||
| Median | 27.7 | |
| Range | 10.1-131.5 | |
| Baseline platelets, × 103/μL | ||
| Median | 27 | |
| Range | 7-88 | |
| Missing | 1 | |
| Sex | ||
| Female | 37 | 53 |
| Male | 33 | 47 |
| Hispanic | ||
| Yes | 6 | 9 |
| No | 50 | 71 |
| Unknown | 14 | 20 |
| Race | ||
| White | 58 | 83 |
| African American/black | 7 | 10 |
| Asian | 1 | 1 |
| Native Hawaiian or other Pacific Islander | 1 | 1 |
| Unknown | 3 | 4 |
| ECOG PS | ||
| 0-1 | 50 | 71 |
| ≥2 | 19 | 27 |
| Not reported | 1 | 1 |
| Age category, y | ||
| <60 | 55 | 79 |
| 60+ | 15 | 21 |
| WBC count category, × 103/μL | ||
| 10-19.9 | 27 | 39 |
| 20-49.9 | 28 | 40 |
| ≥50 | 15 | 21 |
| Characteristic | n | % |
|---|---|---|
| Age, y | ||
| Median | 46.5 | |
| Range | 19.1-86.3 | |
| Baseline WBC count, × 103/μL | ||
| Median | 27.7 | |
| Range | 10.1-131.5 | |
| Baseline platelets, × 103/μL | ||
| Median | 27 | |
| Range | 7-88 | |
| Missing | 1 | |
| Sex | ||
| Female | 37 | 53 |
| Male | 33 | 47 |
| Hispanic | ||
| Yes | 6 | 9 |
| No | 50 | 71 |
| Unknown | 14 | 20 |
| Race | ||
| White | 58 | 83 |
| African American/black | 7 | 10 |
| Asian | 1 | 1 |
| Native Hawaiian or other Pacific Islander | 1 | 1 |
| Unknown | 3 | 4 |
| ECOG PS | ||
| 0-1 | 50 | 71 |
| ≥2 | 19 | 27 |
| Not reported | 1 | 1 |
| Age category, y | ||
| <60 | 55 | 79 |
| 60+ | 15 | 21 |
| WBC count category, × 103/μL | ||
| 10-19.9 | 27 | 39 |
| 20-49.9 | 28 | 40 |
| ≥50 | 15 | 21 |
ECOG PS, Eastern Cooperative Oncology Group performance status; WBC, white blood cell.
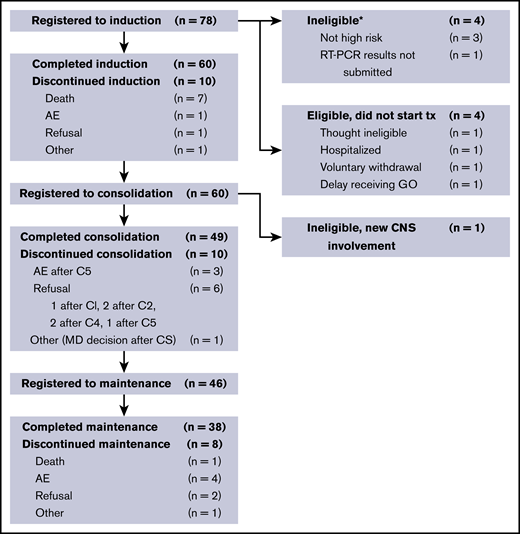
Study flow diagram. Note that 1 patient who achieved CR did not complete induction therapy because of insurance issues. *Two patients who were ineligible for step 1 also registered for steps 2 and 3; they are not shown here after induction therapy. AE, adverse event; C, cycle; CNS, central nervous system; MD, treating physician; RT, reverse transcription; tx, treatment.
Study flow diagram. Note that 1 patient who achieved CR did not complete induction therapy because of insurance issues. *Two patients who were ineligible for step 1 also registered for steps 2 and 3; they are not shown here after induction therapy. AE, adverse event; C, cycle; CNS, central nervous system; MD, treating physician; RT, reverse transcription; tx, treatment.
Sixty patients (86%) achieved a documented CR. Of the 10 nonresponding patients (14%), 7 died before or without having a response assessment, 2 had an inadequate response assessment, and 1 had newly detected central nervous system leukemia after completion of induction therapy. With a median follow-up of 3.4 years, the Kaplan-Meier–determined 3-year EFS estimate was 78% (95% confidence interval [CI], 67%-86%), which was significantly improved compared with the historical, protocol-specified rate of 50% (1-sided P < .001; Figure 3A). Using the binary EFS end points, we found the 3-year EFS to be 74% (95% CI, 62%-84%), which was significantly higher than 50% (1-sided P < .001).
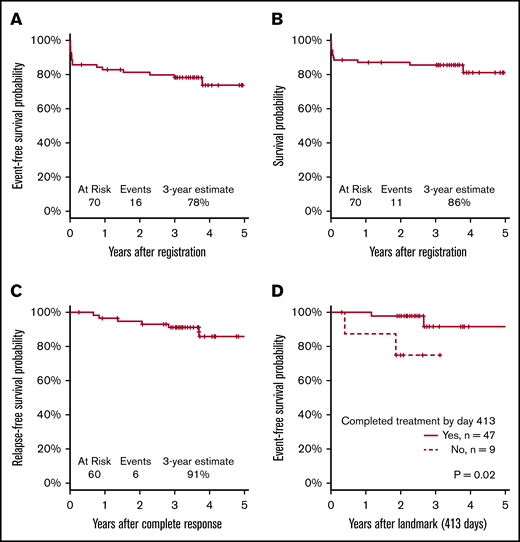
The 3-year OS estimate was 86% (95% CI, 75%-92%; Figure 3B). The 3-year RFS estimate was 91% (95% CI, 80%-96%; Figure 3C). RFS events occurred in 6 patients (10%) at a median of 1.7 years after CR (range, 0.7-3.7 years); these events included 4 documented relapses and 2 deaths during remission. Median time to CR was 0.1 years (39.5 days; range, 18 days to 1.1 years). Because not all patients completed postremission therapy as planned, a landmark analysis of patients who achieved CR was performed. The analysis showed a significant difference in EFS between patients who did or did not complete consolidation therapy (P = .02; Figure 3D). The CIR at 3 years post-CR was 7.1% (95% CI, 2.2% to 15.8%).
Serial molecular response assessment data were available for 37 (62%) of the responding patients. Among the 22 patients with response (postinduction) assessment data available within 8 weeks after diagnosis, 17 (77%) had conversion to PML-RARα−. Among the patients assessed before 8 weeks, those who had a PCR response were less likely to have an EFS event than those who did not (P = .03). All of the 15 patients (100%) whose posttreatment PCR results were unavailable until after 8 weeks had conversion to PML-RARα−.
Safety
Eight patients (11%; 95% CI, 5%-21%) died within 6 weeks of initiating therapy, showing that the 6-week mortality rate was significantly lower than the protocol-specified rate of 30% (1-sided P < .001). Causes of death included hemorrhage (n = 3), infection (n = 2), and hepatic failure (n = 1). Two early deaths were of indeterminate causes.
Grade 3 to 5 toxicities that occurred during induction therapy are shown in Table 2, the most common of which included febrile neutropenia, aspartate aminotransferase/alanine aminotransferase elevation, hyperglycemia, hypoxia, headache, and prolonged QT interval corrected for heart rate. Retinoic acid syndrome occurred in 6 patients (9%), with only 1 case (1%) considered to be grade 3/4. All documented cases of retinoic acid syndrome occurred during the induction phase.
Treatment-emergent grade 3 to 5 adverse events in ≥10% of patients during any phase of therapy
| Adverse event | Induction (N = 70), n (%) | Consolidation (n = 59), n (%) | Maintenance (n = 46), n (%) |
|---|---|---|---|
| Febrile neutropenia | 29 (41) | 31 (53) | 2 (4) |
| Infection | 14 (20) | 14 (24) | 4 (9) |
| Hyperglycemia | 12 (17) | 5 (8) | 5 (11) |
| AST/ALT/GGT/ALP | 11 (16) | 6 (10) | 6 (13) |
| Headache | 10 (14) | 10 (17) | 7 (15) |
| Hypoxia | 10 (14) | 0 (0) | 1 (2) |
| DIC | 8 (11) | 1 (2) | 0 (0) |
| Dyspnea | 8 (11) | 4 (7) | 2 (4) |
| QTc prolonged | 8 (11) | 3 (5) | 0 (0) |
| Hypokalemia | 7 (10) | 4 (7) | 0 (0) |
| Fatigue | 4 (6) | 8 (14) | 4 (9) |
| Pain | 4 (6) | 8 (14) | 2 (4) |
| Nausea | 2 (3) | 7 (12) | 6 (13) |
| Hematologic | |||
| Neutrophils | NA | 9 (20) | |
| Hemoglobin | NA | 3 (7) | |
| Platelets | NA | 3 (7) | |
| Adverse event | Induction (N = 70), n (%) | Consolidation (n = 59), n (%) | Maintenance (n = 46), n (%) |
|---|---|---|---|
| Febrile neutropenia | 29 (41) | 31 (53) | 2 (4) |
| Infection | 14 (20) | 14 (24) | 4 (9) |
| Hyperglycemia | 12 (17) | 5 (8) | 5 (11) |
| AST/ALT/GGT/ALP | 11 (16) | 6 (10) | 6 (13) |
| Headache | 10 (14) | 10 (17) | 7 (15) |
| Hypoxia | 10 (14) | 0 (0) | 1 (2) |
| DIC | 8 (11) | 1 (2) | 0 (0) |
| Dyspnea | 8 (11) | 4 (7) | 2 (4) |
| QTc prolonged | 8 (11) | 3 (5) | 0 (0) |
| Hypokalemia | 7 (10) | 4 (7) | 0 (0) |
| Fatigue | 4 (6) | 8 (14) | 4 (9) |
| Pain | 4 (6) | 8 (14) | 2 (4) |
| Nausea | 2 (3) | 7 (12) | 6 (13) |
| Hematologic | |||
| Neutrophils | NA | 9 (20) | |
| Hemoglobin | NA | 3 (7) | |
| Platelets | NA | 3 (7) | |
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; DIC, disseminated intravascular coagulation; GGT, γ-glutamyl transferase; NA, not applicable; QTc, QT interval corrected for heart rate.
Fifty-nine (98%) of the 60 patients who achieved a CR began consolidation therapy. Of these, 49 (83%) completed all planned consolidation treatments. Reasons for discontinuing consolidation therapy included adverse events and voluntary withdrawal. The most common grade 3 to 5 toxicities during consolidation therapy included febrile neutropenia, fatigue, headache, and nausea.
Forty-six (94%) of the 49 patients who completed consolidation therapy began maintenance therapy. Of these patients, 38 (83%) completed all planned maintenance treatments. One patient died (nonleukemia associated) during the maintenance phase, and 7 other patients came off therapy during the maintenance phase because of adverse events or voluntary withdrawal. The most common grade 3 to 5 toxicities during the maintenance phase included nausea, headache, and alanine aminotransferase elevation. There were no reported cases of venoocclusive disease of the liver. Of the initial 60 patients who achieved a CR, 38 (63%) completed all subsequent phases of therapy.
Discussion
Although APL is generally considered to be highly curable, patients with high-risk APL have historically experienced disproportionately higher rates of both early death and relapse. Earlier trials using chemotherapy plus ATRA combinations, in which ATRA was administered only during the induction phase, yielded CIR rates in the range of 25% to 50% among high-risk patients.9,10 Follow-up trials that added ATRA to postremission therapy demonstrated improved outcomes, even among high-risk patients, although relapse rates in this subgroup remained as high as 21%.11 Subsequent studies that used cytarabine-containing consolidation regimens seemed to have further reduced the CIR among high-risk patients.12,13 Although several recent studies have demonstrated improved outcomes among APL patients treated with ATO-containing regimens during induction and/or consolidation, a minority of patients in these trials had high-risk disease, making it difficult to assess the efficacy of an ATO-containing regimen among this patient subgroup.5,6,14,15
Our clinical trial reported the outcomes of the largest number of high-risk APL patients to be treated with an ATO-containing regimen to date. Although this clinical trial was nonrandomized and was originally designed when relapse rates for high-risk APL patients remained high, the 78% 3-year EFS of this high-risk population was encouraging and compared favorably to other modern regimens, including risk-adapted chemotherapy plus ATRA regimens12,13 and ATO-containing regimens.5,14 In addition, only 4 cases of documented relapse occurred during the course of the study; none occurred during treatment. Given that relapse historically accounts for a high percentage of overall treatment failure among high-risk APL patients, the very low incidence of relapse in this study suggests that our regimen, similar to other modern regimens, may effectively mitigate a key component of treatment failure. Another interesting observation in this study was a high rate of molecular response after induction therapy. EFS seemed superior among patients who achieved early molecular response, suggesting that early molecular response from an ATRA plus ATO plus GO regimen such as ours could be an important end point for high-risk APL patients and is worthy of further study.
Historically, high-risk APL has been associated with an early death rate of ∼20%, with some reports indicating rates near 30%.6,12,13,16 Overall, the induction regimen of ATRA plus ATO plus GO in our study was well tolerated and was associated with a relatively low rate of early death. This seems consistent with other smaller studies of high-risk APL patients using an ATRA plus ATO–based induction regimen.15,17 However, the postremission regimen of our study was quite intensive, and the inclusion of several rounds of consolidation containing anthracyclines and GO reflected the fact that most APL regimens at the time of protocol design included dose-intensive therapies during consolidation, a strategy that is changing in the current era. It is noteworthy that a significant minority (37%) of patients who achieved remission did not complete all planned phases of therapy, including 12% of patients who discontinued treatment during the postremission phase of the regimen (consolidation plus maintenance) because of adverse events. This discontinuation rate suggests that undergoing postremission therapy with cytotoxic drugs such as GO and daunorubicin may be difficult for many patients, an important observation given the fact that EFS seemed to be inferior among patients who achieved a CR without completing all planned postremission treatments. With the evolution of APL therapy toward noncytotoxic regimens, postremission therapy with ATRA plus ATO could be considered a more tolerable and less toxic alternative treatment for high-risk APL patients, similar to treatments that have been used in non–high-risk protocols.3,5,14 To this end, an ongoing study of an ATRA plus ATO–based regimen for high-risk APL patients in Europe uses a less intense postremission strategy that includes ATRA plus ATO only (registered at www.clinicaltrials.gov as #NCT02688140).
In summary, a therapeutic regimen of ATRA plus ATO plus GO for patients with high-risk APL was effective and had a very low risk of relapse and a high rate of early molecular response, suggesting that this regimen could be considered an option for high-risk patients. Future trials that include ATRA plus ATO plus GO induction therapy with a modified, less dose-intensive postremission strategy are warranted.
For original data, please contact the corresponding author at jeffrey.lancet@moffitt.org.
Acknowledgments
Editorial assistance was provided by the Moffitt Cancer Center’s Scientific Editing Department by Paul Fletcher and Daley Drucker. No compensation was given beyond their regular salaries.
This study was supported by National Cancer Institute, National Institutes of Health, grants CA180888, CA180819, CA180820, CA180821, CA189848, CA189808, CA189860, CA180801, CA189953, CA180818, CA233290, CA233320, CA232760, and CA46282, and in part by Wyeth Pharmaceuticals (Pfizer, Inc.).
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: J.E.L. served as the national principal investigator, designed the research, performed the research, participated in data interpretation, and wrote the manuscript; A.B.M. and M.O. performed biostatistical analyses and participated in data interpretation and editing of the manuscript; S.E.C., D.J.D., M.S.T., M.R.L., R.S.K., and H.P.E. served as coinvestigators, performed the research, and participated in data interpretation and editing of the manuscript; and F.R.A. designed the research and participated in data interpretation and editing of the manuscript.
Conflict-of-interest disclosure: J.E.L. has a consulting or advisory role with Jazz Pharmaceuticals, Agios, AbbVie, Pfizer, and Daiichi-Sankyo and receives research funding from Prescient. M.S.T. receives research funding from AbbVie, Cellerant, Orsenix, ADC Therapeutics, and Biosight and has a consulting or advisory role with AbbVie, BioLineRx, Daiichi-Sankyo, Orsenix, KAHR, Rigel, Nohla, Delta-Fly Pharma, Oncolyze, and Jazz Pharmaceuticals. M.O. has a consultant or advisory role with Celgene, Glycomimetics, and Merck. The remaining authors declare no competing financial interests.
Correspondence: Jeffrey E. Lancet, Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, 12902 USF Magnolia Dr, Tampa, FL 33612; e-mail: jeffrey.lancet@moffitt.org.

