

Abstract
Since the discovery of FMS-like tyrosine kinase-3 (FLT3)–activating mutations as genetic drivers in acute myeloid leukemia (AML), investigators have tried to develop tyrosine kinase inhibitors that could effectively target FLT3 and alter the disease trajectory. Giltertinib (formerly known as ASP2215) is a novel compound that entered the field late, but moved through the developmental process with remarkable speed. In many ways, this drug’s rapid development was facilitated by the large body of knowledge gained over the years from efforts to develop other FLT3 inhibitors. Single-agent gilteritinib, a potent and selective oral FLT3 inhibitor, improved the survival of patients with relapsed or refractory FLT3-mutated AML compared with standard chemotherapy. This continues to validate the approach of targeting FLT3 itself and establishes a new backbone for testing combination regimens. This review will frame the preclinical and clinical development of gilteritinib in the context of the lessons learned from its predecessors.
Introduction
On 21 September 2018, gilteritinib (XOSPATA) received marketing approval in Japan for the treatment of relapsed or refractory (R/R) FLT3-mutated acute myeloid leukemia (AML); on 28 November of the same year, the US Food and Drug Administration followed suit, approving gilteritinib for the same indication in the United States. Although to some it seemed as if the drug appeared out of nowhere, gilteritinib’s rapid advance to approval was the result of efficient translation of all that had been learned about the disease and the target, FMS-like tyrosine kinase-3 (FLT3), over the prior 2 decades. Multiple drugs have been studied over the years as FLT3 inhibitors, but most were thwarted by lack of potency, off-target effects, and different mechanisms of resistance. It was as if successful FLT3 inhibition was a golden idol, hidden in a temple laden with deadly pitfalls. The early drug-development teams in this effort rushed into the temple, only to spring (and reveal) all of the traps. The developers of gilteritinib were the explorers who, after carefully observing everyone else’s missteps, strolled in afterward to claim the prize.
In this review, we will first focus on the laboratory science behind gilteritinib’s development and then describe its clinical development, including its current place in AML therapeutics. Finally, we will conclude with future directions for gilteritinib and FLT3 inhibitors, in general.
FLT3 and AML
FLT3-mutated AML
Even 20 years ago, an important negative prognostic factor for AML was known to be a high white blood cell count.1 In retrospect, it seems likely that a large fraction of these patients whose poor prognosis was derived from hyperleukocytosis harbored activating mutations in the FLT3 gene. The most common mutation, discovered in Japan and reported in 1996,2 is the internal tandem duplication (ITD). FLT3 tyrosine kinase domain (FLT3-TKD) mutations were discovered a few years later by 2 independent groups.3,4 The incidence of these 2 mutations varies depending on the patient population being studied. However, in newly diagnosed patients younger than 65 years of age, the overall incidence is probably ∼30%: 23% for FLT3-ITD mutations and 7% for FLT3-TKD mutations.5
FLT3-ITD mutations were quickly recognized as the more troublesome of the 2 classes of activating mutations. Patients with an FLT3-ITD mutation are younger, on average, than the typical AML patient.6 Most achieve remission with conventional induction chemotherapy, but they have a pronounced tendency to relapse, relapse quickly, and die sooner than AML patients of a similar age lacking such a mutation.7,8 The FLT3-TKD mutations are less common and have less prognostic impact at diagnosis, but they are clinically quite important, particularly as a mechanism of resistance to some FLT3 inhibitors.9,10 Early efforts at improving outcomes for patients with FLT3-ITD AML were appropriately focused on consolidation with allogeneic transplant. An aggressive approach to hematopoietic stem cell transplant (HSCT) patients in first remission remains arguably the most important component of therapy.11-14 Nonetheless, HSCT is not a fail-safe approach, because an FLT3-ITD mutation remains an independent risk factor for post-HSCT relapse.15,16 Using higher doses of daunorubicin during induction was of some benefit17,18 ; however, even with high-dose daunorubicin (90 mg/m2 × 3 doses), FLT3-ITD AML patients in the Eastern Cooperative Oncology Group 1900 study showed a cumulative incidence of relapse of 61% and estimated 4-year event-free survival and overall survival (OS) of 23% and 28%, respectively.17 These outcomes suggest much room for improvement. A constitutively activating kinase represents an obvious therapeutic target in oncology, and a FLT3 inhibitor “arms race” quickly developed shortly after the discovery of FLT3-activating mutations.
The target: FLT3
The receptor tyrosine kinase (RTK) FLT3 is a member of the so-called “split kinase” type 3 family of RTKs; as such, it shares homology with KIT, the platelet-derived growth factor receptors, and colony stimulating factor-1 receptor.19 Therefore, FLT3 inhibitors will often inhibit 1 or more of these other family members as well. FLT3’s principal role in hematopoiesis is at the progenitor level, where it drives expansion of different subsets within this compartment (Figure 1). That FLT3 is important in hematopoiesis is well established, but the exact role that it plays in defining specific progenitor cell types continues to be debated.20-23 Debate also continues about how much, if any, of this receptor is expressed at the stem cell level; however, it is clear that as the blood cells mature, most lose expression of FLT3, with the exception of dendritic cells, which remain at least partially dependent on FLT3 for proliferation.24 The impact of FLT3 inhibition on any of the progenitor populations is still unclear, because activation of redundant pathways may compensate for the loss of FLT3 signaling. FLT3 is a cytokine receptor, and its cognate ligand, FLT3 ligand, is expressed rather ubiquitously, albeit at a low level most of the time.25 The level of FLT3 ligand increases by 2 or 3 orders of magnitude during chemotherapy-induced aplasia,26-28 which would be expected to drive the expansion of multipotent progenitor cells to promote hematopoietic recovery. Finally, the dependence of mature dendritic cells (Figure 1) on FLT3 signaling may have implications for patients treated with FLT3 inhibitors. Inhibition of dendritic cell function may result in increased infection risk, particularly in patients who have undergone allogeneic transplant and are receiving an FLT3 inhibitor as maintenance therapy.
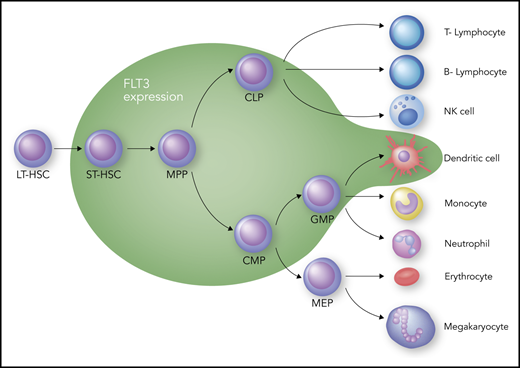
Hematopoiesis and FLT3 expression. The green zone surrounds the cell types that express FLT3. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor; LT-HSC, long-term hematopoietic stem cell; MEP, megakaryocyte-erythroid progenitor; MPP, multipotent progenitor; NK, natural killer; ST-HSC, short-term hematopoietic stem cell. Professional illustration by Somersault18:24.
Hematopoiesis and FLT3 expression. The green zone surrounds the cell types that express FLT3. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor; LT-HSC, long-term hematopoietic stem cell; MEP, megakaryocyte-erythroid progenitor; MPP, multipotent progenitor; NK, natural killer; ST-HSC, short-term hematopoietic stem cell. Professional illustration by Somersault18:24.
An FLT3-ITD mutation consists of a duplication of coding sequence that is inserted in tandem and in-frame.13 The length can vary from 3 bp, coding for a single amino acid, to >200 bp. The duplication almost invariably starts within the juxtamembrane domain, most commonly involving residue arginine 595.29 Often, there is an additional inserted sequence that is unique (but always in-frame), usually coding for a single amino acid. The juxtamembrane domain normally exerts a negative regulatory effect on the kinase activity of FLT3, such that structural perturbations (such as these tandem duplications of juxtamembrane coding sequence) release the receptor from autoinhibition.30 The length is not the only variable component of this mutation: the insertion site can vary, because of the variation in length. A short duplication is usually confined to the juxtamembrane domain coding sequence (Figure 2), whereas a longer duplication extends into the first kinase domain, meaning the insertion site is actually within the coding sequence for the kinase domain. These longer insertions are more problematic to detect (because the sequence itself is normal, just duplicated) and almost certainly have a different biology, likely conferring a more aggressive phenotype.31-33 Patients with longer insertions appear to have decreased benefit from the combination of midostaurin and induction/consolidation chemotherapy.34
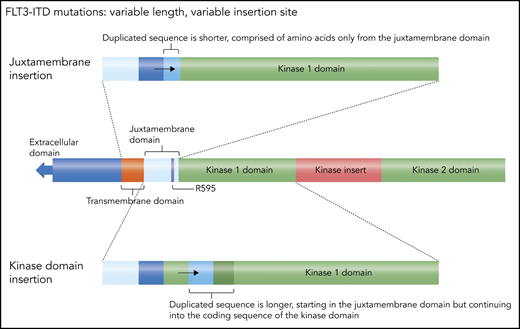
FLT3 ITDs. This diagram illustrates how FLT3 mutations can be defined as “juxtamembrane insertion” and “kinase domain insertion.” For the large majority of FLT3-ITD mutations, the duplicated sequence includes the codon for residue arginine 595 (R595).29 The resultant amino acid sequence inserted typically ranges from 3 to 42 residues. When the insertion is short, it consists only of juxtamembrane sequence, whereas longer duplications include residues from the kinase 1 domain. Therefore, in the case of these longer insertions, the actual duplicated sequence begins within that kinase domain and are sometimes referred to as a kinase domain insertion. Professional illustration by Somersault18:24.
FLT3 ITDs. This diagram illustrates how FLT3 mutations can be defined as “juxtamembrane insertion” and “kinase domain insertion.” For the large majority of FLT3-ITD mutations, the duplicated sequence includes the codon for residue arginine 595 (R595).29 The resultant amino acid sequence inserted typically ranges from 3 to 42 residues. When the insertion is short, it consists only of juxtamembrane sequence, whereas longer duplications include residues from the kinase 1 domain. Therefore, in the case of these longer insertions, the actual duplicated sequence begins within that kinase domain and are sometimes referred to as a kinase domain insertion. Professional illustration by Somersault18:24.
In some of the retrospective studies establishing the negative prognostic effect of FLT3-ITD mutations, it was recognized that the amount of mutant allele relative to the wild-type (nonmutated) allele was important.7,35 The capillary electrophoresis method of separating polymerase chain reaction (PCR) products allowed for the calculation of an allelic ratio (mutant allele/wild-type allele).36 The mutant burden can also be expressed as a variant allele frequency (VAF; amount of mutant/amounts of mutant plus wild-type alleles). Either way, patients with relatively more mutant burden had higher relapse rates and lower survival. Although it is probably safe to conclude that the more FLT3-ITD alleles present, the worse the disease, setting a strict threshold of allelic burden for the purposes of clinical decision making is not very practical at the present time. Current assay methods are not standardized, intrasample variability is high,37 and outcomes may be influenced by chemotherapy and transplant regimens, which vary widely around the world. Standardization of the FLT3-ITD detection assay is an essential future goal for the field.
An important issue pertaining to FLT3-ITD AML is minimal (or measurable) residual disease (MRD). An argument can be made that an FLT3-ITD mutation is not useful as a marker of MRD, because it occurs late in leukemogenesis and can change or be lost at relapse.38,39 However, the unique length of any patient’s insertion mutation provides a signature of that patient’s disease. The greater obstacle for using FLT3-ITD mutations for MRD is the fact that, with conventional PCR, the wild-type allele outcompetes the mutant allele during amplification, a phenomenon referred to as “template bias.”40 Combination PCR-NGS (next-generation sequencing) assays appear to overcome this technical challenge, and such assays will likely yield important information about FLT3-ITD MRD in ongoing studies of FLT3 inhibitors.41-43
Efforts to develop FLT3 inhibitors: a prelude to gilteritinib
After preclinical evaluation of different FLT3 tyrosine kinase inhibitors (TKIs) showed clear evidence of antileukemic activity,44,45 clinical trials of FLT3 inhibitors in AML enrolled their first patients by 2002. Early reports on agents, such as midostaurin, lestaurtinib, and sunitinib, attested to some degree of clinical activity of these drugs, with relatively frequent reports of reduction or elimination of circulating blasts and modest extramedullary toxicity.46-48 Because these trials were plagued by variable and often low numbers of enrolled patients with FLT3 mutations, their ability to use clinical response to demonstrate efficacy and/or facilitate dose optimization was diluted. Additionally, these drugs were known preclinically to have potency against FLT3, as well as many other kinases. The first generation of tested agents was repurposed multikinase-inhibiting drugs and, to a large extent, was “FLT3 inhibitors” more by accident than design.
Still, some dramatic patient responses suggested that these drugs had potential for clinical activity. However, the relatively common finding of rapid and profound reduction or elimination of circulating blasts generally was not paired with reductions in marrow blast content, and antileukemic effects usually became lost within a few weeks. Few, if any, of these responses provided sufficient disease stabilization to consider allogeneic transplant and so the clinical relevance of responses to first-generation FLT3 inhibitors as single agents was unclear. Dose escalation in an attempt to improve response generally only increased bothersome side effects, such as nausea, diarrhea, asthenia, or hand-foot syndrome.48-50 Moreover, these drugs indeed showed antileukemic activity in patients with FLT3-mutated and FLT3 wild-type AML, which raised the question of whether the observed clinical activity was due to FLT3 inhibition at all.
Therefore, despite a relatively small sample size of patients with R/R AML studied, none of the first several FLT3 inhibitors emerged as a strong contender for single-agent use to treat clinically active leukemia. This left the field wondering: Was the failure of first-generation early FLT3 inhibitors as single agents due to suboptimal drugs, a bad target, or a little of both? The way to answer this question was to develop drugs that were more potent and selective inhibitors of FLT3, with which target validation could follow.
A second generation of inhibitors: some problems solved, but new issues emerge
Quizartinib (formerly AC220) was the first drug specifically developed as a potent selective FLT3 inhibitor for AML. First identified from an in vitro screening method from which the current commercial KINOMEscan assay is derived, quizartinib showed high selectivity at low concentrations that was almost entirely limited to FLT3, KIT, colony-stimulating factor-1 receptor, platelet-derived growth factor receptor, and RET kinase.51,52 Plasma inhibitory activity (PIA) assay, an ex vivo pharmacodynamics test that estimates the potential for FLT3 inhibition in in vivo testing,53 showed exquisitely potent activity against FLT3 that was objectively superior to all previously tested agents and was consistently observed at all tested doses during phase 1. Now one could examine whether a “real” FLT3 inhibitor would generate “real” responses. The answer turned out to be “yes” and “no.”
Unlike prior FLT3 inhibitors, responses to quizartinib in patients with R/R FLT3-ITD AML paired near-universal clearance of peripheral blasts with frequent reduction, if not complete elimination, of marrow blasts. Quizartinib responses occurred primarily in patients with FLT3-ITD mutations and occurred at low doses (as little as 18 mg daily).54-56
Marrow responses to quizartinib differed from responses seen with traditional cytotoxic chemotherapy. Peripheral blast clearance occurred in only a few days, but marrow blast clearance usually required ≥4 weeks of therapy and often was achieved without intervening marrow aplasia. Interestingly, responding patients frequently, although not universally, showed persistence of the FLT3-ITD mutation in recuperating marrow cells and circulating neutrophils at hematologic reconstitution, suggesting that induction of terminal differentiation was a mechanism of response for a substantial subset of patients.57,58 Full criteria for complete remission (CR) were seen only rarely in these trials, and significant and persistent cytopenias during response were common and generally did not resolve with dose reduction or interruptions. Although potent KIT inhibition by quizartinib almost certainly contributed to ongoing transfusion burden,59 persistence of granulocytic hyperplasia by differentiating cells during response may also have contributed.
Regardless of these issues, quizartinib remained active, tolerable, and frequently allowed subsequent transplant (32%-37% of enrolled patients),60 all of which rekindled interest in clinical targeting of FLT3. Amplifying this interest was the demonstration that midostaurin added to frontline induction improved survival compared with placebo, prompting the first drug approval of a TKI in patients with FLT3-mutated AML for combination therapy.61 With minor dosing refinement, quizartinib appeared poised to take FLT3-targeted therapy to a higher level. However, a major limitation emerged: resistance due to on-target kinase-activating FLT3 mutations, which often occurred within 2 to 4 months of initiating quizartinib.9 The clinical confirmation of drug resistance driven by reactivation of FLT3 kinase activity argued strongly for drugs capable of potent FLT3 inhibition but with less susceptibility to second-site FLT3 mutations that conferred resistance (meaning a type I inhibitor, as discussed in "Lesson 5: FLT3-TKD mutations confer resistance to type II FLT3 inhibitors").62-64 In large part because of the frequency of QT prolongation, quizartinib’s regulatory development slowed, and multiple phase 2 trials were required to clarify its optimal dose (ultimately settling on 30-60 mg for single-agent use) before pivotal trials could be launched.
The lessons learned from prior FLT3 inhibitors
A long list of compounds were studied as FLT3 inhibitors in the laboratory and carried into multiple clinical trials: midostaurin, lestaurtinib, sorafenib, sunitinib, tandutinib, quizartinib, and crenolanib, to name just some.45-49,52,54,62,65-69 Although it certainly seemed like the failures outweighed the successes, each failure and each success produced lessons that ultimately led to the development of gilteritinib.70
Lesson 1: inhibition of FLT3 needs to be near complete.
Although FLT3 triggers phosphorylation and activation of key signaling proteins, such as STAT5 and MAPK, autophosphorylation of the FLT3 receptor itself is an excellent biomarker for its activation, and loss of that autophosphorylation signifies successful inhibition. The degree of phosphorylation can be quantified, using immunoblotting or enzyme-linked immunosorbent assay–based techniques, directly in circulating blasts or via a PIA assay (Figure 3) using the patient’s plasma.53 Because of the high degree of plasma protein binding of these generally hydrophobic drugs, estimates of drug potency using in vitro cell culture models routinely underestimate (by 1 or 2 orders of magnitude) the concentrations necessary to inhibit the target in human patients. Early trials indicated that clinical activity of FLT3 TKIs correlated relatively tightly with inhibition of FLT3 autophosphorylation to <15% of baseline, and even lower if possible.47,53
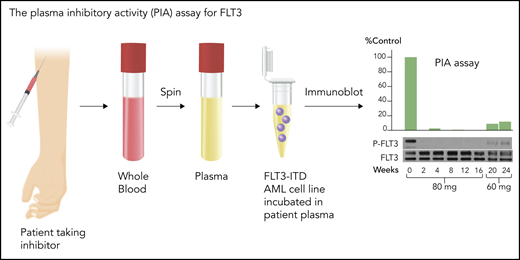
PIA assay for FLT3. This assay serves as a validated surrogate for in vivo FLT3 inhibition in patients treated with FLT3 inhibitors. Whole blood is collected at a trough time point from patients treated with an FLT3 inhibitor, preferably when the drug is at steady-state. An FLT3-ITD–expressing cell line, such as Molm14, is then incubated in the plasma, and the phosphorylation status of FLT3 is analyzed by immunoblotting. The results are normalized to control or pretreatment plasma. The immunoblots can be quantitated by densitometry. The blots are from a patient treated with 80 mg/d or 60 mg/d of lestaurtinib, a first-generation FLT3 inhibitor.103 Professional illustration by Somersault18:24.
PIA assay for FLT3. This assay serves as a validated surrogate for in vivo FLT3 inhibition in patients treated with FLT3 inhibitors. Whole blood is collected at a trough time point from patients treated with an FLT3 inhibitor, preferably when the drug is at steady-state. An FLT3-ITD–expressing cell line, such as Molm14, is then incubated in the plasma, and the phosphorylation status of FLT3 is analyzed by immunoblotting. The results are normalized to control or pretreatment plasma. The immunoblots can be quantitated by densitometry. The blots are from a patient treated with 80 mg/d or 60 mg/d of lestaurtinib, a first-generation FLT3 inhibitor.103 Professional illustration by Somersault18:24.
Lesson 2: inhibition of FLT3 needs to be sustained for days, not hours.
This may seem intuitive, but the duration of inhibition of any kinase inhibitor in vivo often is not assessed in clinical trials. With FLT3, inhibition lasting only a few hours each day is insufficient to kill FLT3-ITD AML cells in vitro or in vivo.71 Therefore, the ideal FLT3 inhibitor is a drug with a long plasma half-life. The dosing interval for crenolanib, for example, had to be increased to 3 times daily to account for its short in vivo half-life.62
Lesson 3: inhibition of c-Kit results in myelosuppression.
Although FLT3 clearly plays an important role in hematopoiesis (Figure 1), transgenic mice deficient in FLT3 develop relatively normally.72 Their hematopoietic stem cells have reduced repopulating efficiency, and their marrow has reduced numbers of lymphoid progenitor cells. In contrast, complete loss of KIT or of its cognate ligand, steel factor/stem cell factor/kit ligand, is embryonically lethal.73 Because of the close structural similarity between FLT3 and KIT, an inhibitor of 1 has the potential to be an inhibitor of the other. FLT3 inhibitors with activity against KIT are expected to be myelosuppressive to some degree. It is not clear whether this is due to complete inhibition of FLT3 combined with partial inhibition of KIT or simply to KIT inhibition alone. However, this has been an impediment for the development of quizartinib, a potent FLT3 inhibitor with activity against KIT.59,74
Lesson 4: FLT3-ITD AML evolves from diagnosis to relapse.
AML is polyclonal from diagnosis, but in vitro studies suggest that only a fraction of the leukemia cells in a newly diagnosed patient are dependent on FLT3-ITD signaling for survival and proliferation. At relapse, a larger proportion of the leukemia population appears to respond to FLT3 inhibition.75 The prediction of this laboratory observation is that treatment of a newly diagnosed patient with FLT3 inhibition as monotherapy would be mostly ineffective, although this has not been tested clinically. Presently, the use of FLT3 inhibitors in newly diagnosed patients is directed at augmenting the effect of chemotherapy (via synergistic cytotoxicity76 ) and preventing the outgrowth of FLT3-driven clones.
Lesson 5: FLT3-TKD mutations confer resistance to type II FLT3 inhibitors.
Data from X-ray crystallography and structural modeling studies of FLT3 and other tyrosine kinases provide support for a model of TKIs (including FLT3 inhibitors) that classifies them into 2 broad categories: type I and type II.77 Type I inhibitors bind to the adenosine triphosphate (ATP) binding site in the active pocket of the enzyme. Much of their specificity is derived from moieties that interact with amino acid residues surrounding the active site. A type II inhibitor (imatinib being a prototypical example) binds to a hydrophobic pocket adjacent to the ATP site. The pocket is not accessible unless the activation loop of the kinase is in the “DFG-out” position (DFG refers to the 3 highly conserved amino acid residues in the activation loop, aspartate, phenylalanine, and glycine), folded against the kinase domain, and blocking access to the substrate. This conformation also results in the phenylalanine of the DFG motif inserted into the hydrophobic groove. A type II inhibitor, such as quizartinib (Figure 4), actually binds in allosteric fashion (eg, by induced fit), sliding in between the phenyl rings of the gatekeeper residue and the DFG motif.78 A kinase domain mutation at position D835 (typically the negatively charged aspartate is replaced with a hydrophobic residue) disrupts the inactive conformation, closing access to the hydrophobic pocket. In addition, a mutation at the gatekeeper residue (F691) disrupts the hydrophobic pocket and blocks the binding of these type II inhibitors. In the relapsed setting, treatment of FLT3-ITD AML with a type II inhibitor, such as quizartinib or sorafenib, will often result in the emergence of a resistance-conferring TKD mutation (which was often present at low levels prior to the start of therapy).9,79 In accordance with this model, gilteritinib, as a type I inhibitor, is unaffected by mutations at D835 and relatively less affected by gatekeeper mutations.64,80
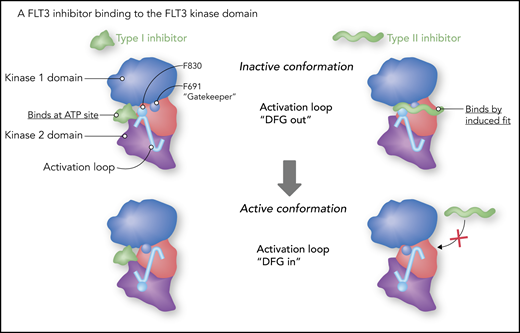
Type I vs type II FLT3 inhibitors. Gilteritinib is a type I inhibitor. As such, it is more or less an ATP mimetic, and its binding is relatively less influenced by the conformation of the activation loop. “DFG” refers to 3 highly conserved amino acid residues (aspartate-phenylalanine-glycine) at the start of the activation loop. A type II inhibitor fits into a hydrophobic groove adjacent to the ATP binding site, and, in the case of quizartinib, actually fits in between phenyl rings of phenylalanine 691 (F691, the “gatekeeper”) and phenylalanine 830 (F830). Professional illustration by Somersault18:24.
Type I vs type II FLT3 inhibitors. Gilteritinib is a type I inhibitor. As such, it is more or less an ATP mimetic, and its binding is relatively less influenced by the conformation of the activation loop. “DFG” refers to 3 highly conserved amino acid residues (aspartate-phenylalanine-glycine) at the start of the activation loop. A type II inhibitor fits into a hydrophobic groove adjacent to the ATP binding site, and, in the case of quizartinib, actually fits in between phenyl rings of phenylalanine 691 (F691, the “gatekeeper”) and phenylalanine 830 (F830). Professional illustration by Somersault18:24.
Lesson 6: treatment of FLT3-ITD AML cells with a FLT3 inhibitor results in a combination of apoptosis and terminal myeloid differentiation.
Early work on first-generation inhibitors indicated that FLT3 inhibition could overcome the block in differentiation in AML cells.81 As FLT3 inhibitors began moving into early clinical trials, translational studies revealed that this phenomenon was occurring in vivo. Sustained potent FLT3 inhibition is quite effective at inducing apoptosis in FLT3-ITD AML cells, provided that they are in suspension culture or circulating outside of the bone marrow microenvironment. However, when these cells are cocultured with bone marrow stromal cells, they differentiate into neutrophils. Within the marrow of a patient treated with FLT3 inhibition, the blasts undergo a mixture of apoptosis and cell cycle arrest, with a large fraction of blasts undergoing terminal differentiation.57,82 In general, FLT3 inhibition as monotherapy results in persistence of disease, which is probably the reason why most patients fail to achieve anything more than a complete response with incomplete count recovery (CRi), a “response” characterized by reduced marrow blasts but incomplete recovery of normal hematopoiesis (often with ongoing transfusion dependence). As a result, responses to FLT3 inhibitors can be difficult to characterize. Bone marrow blasts may be reduced in absolute numbers, but the FLT3-ITD mutation is still routinely detected, often at the same allelic burden as at the start of therapy.57,83 In fact, FLT3 inhibitors can induce a differentiation syndrome that resembles that seen in acute promyelocytic leukemia or isocitrate dehydrogenase (IDH)–mutated AML treated with IDH inhibitors.84,85
Lesson 7: FLT3 inhibitors need to be well tolerated by patients.
Given that these drugs are administered often over months or years, a favorable side effect profile is a crucial component for a TKI. Drugs that cause side effects, such as nausea (and require pretreatment with antiemetics) or hand-foot syndrome (a notorious problem with sorafenib), lower quality of life and are invariably associated with increased rates of noncompliance.86
To summarize, the ideal FLT3 inhibitor should be very well tolerated to take on a daily basis. It should be potent and have a long half-life so that it can completely inhibit FLT3 autophosphorylation in vivo in a sustained fashion. It should not inhibit KIT, which would exacerbate the myelosuppression already present in these patients, but it should be able to inhibit FLT3-TKD mutations to delay the emergence of resistance. Its use in a patient with R/R FLT3-ITD AML will result in clearance of circulating blasts and differentiation of marrow blasts.
Enter gilteritinib.
Gilteritinib: preclinical studies
Gilteritinib is a type I inhibitor
Gilteritinib is a pyrazine carboxamide derivative synthesized and developed by Astellas Pharma, Inc. (Tokyo, Japan) (Figure 5).80 Computational modeling studies suggest that the drug binds to the ATP binding site, with some interaction with the F691 gatekeeper residue. It is a type I inhibitor, generally unaffected by mutations in the activation loop (eg, at D835). In vitro kinase assays using the intracellular domains of different kinases (expressed as a GST-fusion protein to facilitate purification) indicate that gilteritinib has activity against FLT3, ALK, and AXL (among other kinases). Cell-based assays almost always provide a better estimate of the activity of a drug in vivo. Using a variety of AML cell culture models (MV4-11, Molm13/14, SEMK2, and Ba/F3-transfectants), the IC50 (concentration at which 50% of the activity is inhibited) of gilteritinib (in cell culture medium with 10% fetal bovine serum) for inhibition of the autophosphorylation of the FLT3-ITD receptor is ∼1 nM, and it is roughly 5 nM for the wild-type FLT3 receptor.64,80 This degree of potency was confirmed against primary patient FLT3-ITD AML blasts cultured in vitro.

Gilteritinib is multitargeted, but it inhibits FLT3 much more than other kinases
There are several approaches to determining a TKI’s selectivity and potency. In vitro kinase assays can be useful as a broad screen for the activity of any inhibitor against the kinases found in the human genome, and numerous commercially available ones are used by pharma companies for this purpose. In general, they use of a panel of kinases, most often GST-fusion proteins, expressed from baculovirus systems. For transmembrane receptors (like FLT3), only the cytoplasmic portion is expressed and used in the assay, and the kinase substrate is often quite artificial (such as a peptide sequence on an enzyme-linked immunosorbent assay plate). These characteristics account for the often significant discrepancies between in vitro kinase assay results and what is actually inhibited by the drug in patients. These assays are no substitute for the assessment of inhibition within the target cell type but often provide a means of comparing different drugs for relative selectivity. In doing so, it is important to use the same assay. An alternate approach is the KINOMEscan platform (Eurofins, San Diego, CA), which determines a drug’s ability to compete with staurosporine (or a similar ligand) for binding to a kinase expressed by a bacteriophage. Therefore, it is not a kinase assay but rather a drug-competition assay, and this relative simplicity may actually provide a more accurate method of comparing different drugs.87 However, when interpreting a KINOMEscan assay, it is important to be aware of the concentration of drug used in the assay. Preferably, it should approximate what is achieved at steady-state in patients on the drug, accounting for differences in plasma protein binding relative to the assay conditions. As analyzed by this assay system (Figure 6), gilteritinib is multitargeted, but importantly, it binds to the mutant versions of FLT3 more effectively than to the wild-type FLT3. This, coupled with its inactivity against KIT, probably accounts for it being relatively less myelosuppressive than other FLT3 inhibitors. Consistent with its status as a type I inhibitor, it displays excellent binding activity to FLT3-TKD mutants and somewhat less activity against the F691L gatekeeper.
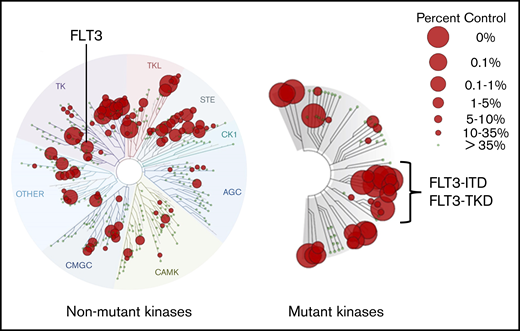
KINOMEscan assay profile for gilteritinib at 100 nM. This commercially available assay of a kinase inhibitor’s relative potency and selectivity is widely displayed but often misunderstood. This profile displays gilteritinib’s affinity for the different kinases in the human genome at 100 nM. A concentration of 100 nM gilteritinib is a crude approximation of steady-state levels achieved in plasma by patients receiving the standard dose of 120 mg/d. The KINOMEscan assay measures the ability of a test compound, such as gilteritinib, at specific concentrations (100 nM in this case) to displace a reference ligand from the active site of a kinase. Therefore, it is not an in vitro kinase assay but rather a drug-binding assay. The reference ligand is a proprietary compound designed to bind in highly promiscuous fashion to kinase active sites (earlier developmental versions of this assay used staurosporine). Kinases, derived from a library of 468 genes, are individually expressed in bacteriophages for use in the assay. In the case of transmembrane receptor kinases, just the cytoplasmic domain is expressed. The library includes the vast majority of wild-type human kinases, as well as key mutant kinases, such as FLT3-ITD and FLT3-TKD variants, including the F691L gatekeeper mutation. Each small gray circle on a branch represents an individual kinase. A red circle over any point of a branch indicates that gilteritinib competes with the reference compound for binding. “Percent control” refers to the efficacy of the competition. The larger the circle, the more effectively it displaces the reference ligand. A value of “0%” (the largest red circle) indicates the highest-affinity binding, meaning no reference ligand binds to the kinase. Note that these are not dissociation constant (Kd) values, although this assay can be used to calculate such values. Images generated using the TREEspot Software Tool and reprinted with permission from KINOMEscan, a division of DiscoveRx Corporation. © 2010 DiscoveRx Corporation.
KINOMEscan assay profile for gilteritinib at 100 nM. This commercially available assay of a kinase inhibitor’s relative potency and selectivity is widely displayed but often misunderstood. This profile displays gilteritinib’s affinity for the different kinases in the human genome at 100 nM. A concentration of 100 nM gilteritinib is a crude approximation of steady-state levels achieved in plasma by patients receiving the standard dose of 120 mg/d. The KINOMEscan assay measures the ability of a test compound, such as gilteritinib, at specific concentrations (100 nM in this case) to displace a reference ligand from the active site of a kinase. Therefore, it is not an in vitro kinase assay but rather a drug-binding assay. The reference ligand is a proprietary compound designed to bind in highly promiscuous fashion to kinase active sites (earlier developmental versions of this assay used staurosporine). Kinases, derived from a library of 468 genes, are individually expressed in bacteriophages for use in the assay. In the case of transmembrane receptor kinases, just the cytoplasmic domain is expressed. The library includes the vast majority of wild-type human kinases, as well as key mutant kinases, such as FLT3-ITD and FLT3-TKD variants, including the F691L gatekeeper mutation. Each small gray circle on a branch represents an individual kinase. A red circle over any point of a branch indicates that gilteritinib competes with the reference compound for binding. “Percent control” refers to the efficacy of the competition. The larger the circle, the more effectively it displaces the reference ligand. A value of “0%” (the largest red circle) indicates the highest-affinity binding, meaning no reference ligand binds to the kinase. Note that these are not dissociation constant (Kd) values, although this assay can be used to calculate such values. Images generated using the TREEspot Software Tool and reprinted with permission from KINOMEscan, a division of DiscoveRx Corporation. © 2010 DiscoveRx Corporation.
Gilteritinib has inhibitory activity against AXL
AXL is a receptor tyrosine kinase of the Tyro3-Axl-Mer family, which carries out a variety of homeostatic functions in multiple tissues.88 These receptors bind membrane-associated ligands, such as Gas6 and protein S. There is some evidence that AXL activation contributes to chemoresistance in AML and specifically contributes to FLT3 activation and response or resistance to FLT3 inhibition.89-91 Although gilteritinib has been touted as an AXL inhibitor, the IC50 against AXL is considerably higher (41 nM) than against FLT3 in cell-based assays; therefore, it is unclear how much of a role, if any, that AXL inhibition plays in any responses to gilteritinib.64
Gilteritinib is relatively nonmyelosuppressive
FLT3 and KIT are 2 structurally related RTKs that play key roles in hematopoiesis.25 The IC50 of gilteritinib for c-Kit is roughly 100 nM (from cell-based assays), 2 orders of magnitude greater than against the FLT3-ITD receptor.64 As noted above, like most FLT3 inhibitors, gilteritinib is ∼fivefold less potent against wild-type FLT3 in comparison with the ITD-mutated version.64 Gilteritinib’s weaker inhibition of wild-type FLT3, combined with its lack of activity against KIT, predicts for a lack of activity against AML cells harboring wild-type FLT3, as well as for a lower degree of myelosuppression relative to other FLT3 inhibitors. The data from bone marrow progenitor cell assays, in which FLT3-ITD inhibitory concentrations of gilteritinib had little effect on normal erythroid and myeloid colony growth, corroborate these predictions.64 However, given the role that FLT3 appears to play in early hematopoiesis (Figure 1), some degree of myelosuppression might be expected with any FLT3 inhibitor.
Gilteritinib is active against FLT3 with TKD mutations
FLT3-TKD mutations are found in roughly 7% of newly diagnosed AML patients, although they do not confer as negative an effect on prognosis as do the ITD mutations.8 At relapse, FLT3-TKD mutations are often lost, but they can still be present and occasionally seem to act as drivers (ie, the disease is highly proliferative, and the FLT3-TKD VAF is high). Gilteritinib has excellent inhibitory activity against all of the FLT3 D835 variants (expressed in Ba/F3 cells),64,80 and it probably will be an effective monotherapy for at least a subset of patients with this mutation subtype. Further studies will be needed to identify which additional clinical and molecular features predict for response of FLT3-TKD AML to gilteritinib.
Gilteritinib is effective against FLT3-TKD mutations that confer resistance to type II inhibitors
Type II FLT3 inhibitors, such as sorafenib and quizartinib, were the first drugs to show significant clinical activity in R/R FLT3-ITD AML,54,69 but they have little or no activity against FLT3-TKD mutations. Genetic analysis of single cells isolated from AML patients suggest, not surprisingly, that these mutations are likely present at subclinical levels at relapse.79 These FLT3-TKD clones, usually at residue D835, emerge under the selective pressure of a type II inhibitor and result in (often very rapid) disease progression.9 In fact, this phenomenon represented important evidence that FLT3 was an important driver mutation in AML. Gilteritinib inhibited FLT3 autophosphorylation and induced a robust cytotoxic effect in vitro against blasts isolated directly from patients developing resistance to sorafenib and quizartinib.64 In vitro, gilteritinib inhibitory activity against FLT3 is not affected by any of the mutations at D835, which essentially eliminates this mode of resistance to therapy. It is important to note that, consistent with the predictions from the structural modeling,80 gilteritinib is significantly less effective at inhibiting FLT3 in the presence of a mutation at the gatekeeper residue (F691).64 This would be a predicted mode of resistance to gilteritinib. In addition, earlier work with other type I inhibitors suggests that activation of downstream or parallel pathways, such as with a RAS mutation,92 would likely confer resistance to gilteritinib, a prediction that is being confirmed in clinical studies.
Gilteritinib potently inhibits FLT3-ITD in vivo
In immunodeficient mice xenografted with MV4-11 AML cells (FLT3-ITD mutated), oral gilteritnib inhibited intratumoral FLT3 autophosphorylation and led to tumor shrinkage.80 Likewise, in mice xenografted with Ba/F3 constructs of FLT3-ITD, FLT3-TKD, and FLT3-ITD-TKD, treatment with oral gilteritinib induced tumor regression. Finally, PIA analysis of plasma samples collected at steady-state from AML patients treated with oral gilteritinib revealed complete suppression of FLT3 autophosphorylation, indicative of sustained high-level FLT3 inhibition in vivo.64
Gilteritinib: clinical studies
A first-in-human study followed that sought to rapidly identify a range of biologically and/or clinically active and tolerable doses. In this manner, lessons learned from the relatively slow development of quizartinib were applied to springboard gilteritinib as quickly as possible from first-in-human testing to phase 3 testing.
The Chrysalis trial was an accelerated-titration phase 1 trial that included multiple phase 2 expansion cohorts.93 Dose-escalation cohorts initially enrolled a single patient who, once safely cleared of the 28-day dose-limiting toxicity window, enabled enrollment of 2 to 5 additional subjects to clarify toxicity profile and expand pharmacokinetics and pharmacodynamics (PIA) datasets at that dose. Concurrently, a higher-dose cohort was opened to a single patient for toxicity evaluation, and the process repeated itself until a maximally tolerated dose was declared. Concurrent with dose escalation, cohort expansion for efficacy assessment was triggered in any dose, as well as at all higher dose levels, that showed potent FLT3 target inhibition by PIA or any composite CR response (CRc) of CR, complete remission with incomplete platelet recovery (CRp), or CRi, defined identically to quizartinib studies. Expansion cohorts were enriched for patients with FLT3 mutations (≥10 of the planned 14 subjects) and could be expanded to a large number of patients (∼60) to refine toxicity and efficacy estimates. This design supported phase 3 power calculations straight from first-in-human results.
Because the first subject in the Chrysalis trial who cleared the dose-limiting toxicity window (20 mg daily, the lowest tested dose) also entered a full CR after 28 days of therapy, all subsequent doses were explored for efficacy once safety was established. Dose-limiting toxicities of gilteritinib occurred at 450 mg daily and were grade 3 diarrhea and liver transaminase elevation, experienced by 1 patient each.93 This left 300 mg as the maximally tolerated dose. Commonly reported toxicities were typical of studies conducted in advanced AML and included cytopenias, febrile neutropenia, and infections, such as pneumonia or sepsis, whereas laboratory abnormalities, including transaminase or creatine phosphokinase elevation, occurred with regularity but rarely were symptomatic or required dose modification to manage. Notably, no patient experienced hand-foot syndrome, grade ≥ 3 QT prolongation was uncommon (3%) and not clearly dose dependent, and mucositis was rare and largely related to hydroxyurea use at screening.
Clinical activity of gilteritinib was observed at the 20 to 40–mg dosing levels but was relatively uncommon, which presumably related to inconsistent, but sometimes potent, FLT3 inhibition at these doses. However, at ≥80 mg, essentially all patients showed profound and continuous FLT3 inhibition, approaching levels seen previously with quizartinib. At these gilteritinib doses, response rates among patients with R/R FLT3-mutated AML (nearly all of whom were FLT3-ITD) reached a plateau, and the CRc rate was 41%. Interestingly, unlike quizartinib trials, CR and CRp, which required transfusion independence, occurred in 16% of gilteritinib-treated patients, with only a slightly higher percentage achieving CRi (24%). Responses were seen in patients who had both FLT3-ITD and D835 mutation, including those who developed FLT3-D835 at progression to sorafenib or quizartinib, and occurred with similar frequency (CRc, 7/13; 55% at ≥80 mg) to those with FLT3-ITD alone (CRc, 77/141; 55%). A substantial number of patients were able to proceed to allogeneic transplant after gilteritinib. From this experience, optimal dosing of 120 mg starting dose, with escalation to 200 mg for lack of CRc, was planned for a phase 3 study as first salvage of FLT3mut+ AML with a comparator group of investigator’s choice of salvage chemotherapy (SC).
The pivotal ADMIRAL study of gilteritinib followed and enrolled 371 patients with FLT3-mutated AML in untreated first relapse after prior remission or who were refractory to frontline induction.94 Patients were randomized 2:1 to single-agent gilteritinib or the investigator’s prerandomization choice of the most appropriate SC regimen from a list of 4 possible options. Of these, 2 were multiagent intensive regimens (mitoxantrone, etoposide, cytarabine or fludarabine or cytarabine, idarubicin, filgrastim) and 2 were single-agent low-intensity regimens (azacitidine or low-dose cytarabine). Responding patients in the gilteritinib arm who proceeded to transplant and remained in remission after successful engraftment could resume gilteritinib as ongoing maintenance therapy. The primary end point was OS and followed an intent-to-treat analysis. During the study, a second coprimary end point, the combined rate of CR and complete remission with partial hematologic recovery (CRh), was added in the gilteritinib arm only. The interim analysis was used to support US regulatory submission but did not alter the trial conduct or analysis.
The ADMIRAL study population had predominantly FLT3-ITD AML (88% FLT3-ITD+ only, 8% FLT3-D835 TKD mutation only, and 2% both mutations). Of keen interest to many was the response rate in patients with only FLT3-TKD mutations. Although there were relatively few such patients, the CR rate in that group (19%) was remarkably similar to the rate in patients with only an FLT3-ITD mutation.94 Sixty-one percent of enrolled patients were relapsed after prior remission, and 39% had refractory disease after induction. Prior therapy included anthracycline in 82% of patients, 20% had received prior HSCT (ie, 32% of relapsed patients), and 12% of patients had received prior midostaurin or sorafenib. Sixty percent of enrolled patients in each arm selected intensive chemotherapy prior to randomization. Median duration of therapy was considerably longer on the gilteritinib arm than on the SC arm (4.1 vs 0.9 cycles).
At interim analysis, the rate of CR and CRh (21%) in the gilteritinib arm was sufficient to prompt drug approval of gilteritinib on 28 November 2018. The label was later expanded based upon final trial results.94 Gilteritinib was associated with higher response rates (CRc rates of 54% for gilteritinib vs 22% for SC) and a statistically significantly longer survival than SC (9.3 months for gilteritinib vs 5.6 months for SC, hazard ratio, 0.64; P < .001, 2-sided log-rank test). CR and CRh rates at the final analysis were 34% and 15%, respectively. Transplant rates were also higher in the gilteritinib arm (26% vs 15%, respectively), and posttransplant gilteritinib therapy was resumed in the majority of transplanted patients. Common toxicities on the gilteritinib arm were cytopenias, infections, and liver transaminase elevations. Hepatic toxicities were predominantly grade 1-2 and only rarely resulted in discontinuation of therapy. Importantly, when corrected for the longer duration of therapy, the rates of grade 3 and higher adverse events were lower in the gilteritinib arm.
Although median survival on the ADMIRAL trial was longer in the gilteritinib arm, the observed gains were largely short term, and no obvious difference in the frequency of long-term surviving patients was seen in comparison with SC. Although median survival remained highly statistically superior in the gilteritinib arm when results were censored for transplant, it is notable that transplanted patients survived longer than nontransplanted patients, and the longest survival on the trial was seen among those who restarted gilteritinib as post-HSCT maintenance. These data demonstrate the ongoing importance of HSCT in the management of R/R AML patients in the era of targeted therapy. They also highlight a potentially important role for ongoing FLT3-targeted therapy to maximize efficacy of transplant following gilteritinib salvage, as well as the need to successfully target or otherwise circumvent gilteritinib-resistance mechanisms.
Overall, therapy with gilteritinib was more effective and less toxic than standard chemotherapy, establishing a new standard of care for management of R/R FLT3-mutated AML. Indeed, gilteritinib occupies a unique position in R/R AML therapy in that it is the only therapy whose US Food and Drug Administration approval for this population was based upon proof of superiority to existing therapy. Finally, it also facilitates outpatient-based therapy of patients, joining other novel agents, such as IDH inhibitors, glasdegib, and venetoclax, that increasingly are positioning AML therapy in this setting.95-98
Understanding responses to gilteritinib
As described above for quizartinib, peripheral blasts clear in response to gilteritinib over a few days, but marrow responses occur slowly, over weeks to a few months. Although marrow hypocellularity/aplasia during gilteritinib is uncommon, response typically is associated with profound cytopenias during the first 1 to 2 months of therapy, especially when hydroxyurea or other cytoreducing agents are used to stabilize relapse prior to gilteritinib initiation. Neutrophil recovery among responding patients typically occurs after 3 to 8 weeks of therapy and may be fastest among patients who differentiate their dominant leukemic clone to gilteritinib. Subsequent transfusion independence occurs in a subset of patients, but the timing is quite variable. Some patients simply do not recover transfusion independence, despite ongoing evidence of no circulating or marrow blasts and no evidence of marrow hypocellularity to suggest typical drug-induced myelosuppression as a cause.
Similar to IDH inhibitors, response to a FLT3 inhibitor can be associated with dramatic differentiation of the leukemic clone, usually in the second month of therapy. Although clinically serious complications of differentiation appear to be rare, gilteritinib side effects, such as pleural or pericardial effusion, fluid retention, and Sweet’s syndrome, may all exist on the spectrum of differentiation syndrome. A black box warning regarding differentiation syndrome risk was included in the US gilteritinib approval, including a recommendation for corticosteroids should it be encountered. Similar to IDH inhibitors,85 differentiation syndrome is rare in the first few weeks of therapy and more typically occurs several weeks to a few months into therapy, typically coincident with neutrophil recovery.
Hematopathologists can be surprised by the marrow morphology during gilteritinib response, which may show a near absence of erythroid or megakaryocytic activity in a marrow that is hypercellular and all but entirely filled with left-shifted granulocytic elements. Some pathologists have termed this “clonal granulocytic hyperplasia,” because, at a molecular level, responding marrows typically remain positive for FLT3 mutation, often with no decrease in allele burden by PCR or VAF by NGS for FLT3-ITD and other leukemia-associated mutations. Molecular persistence with FLT3 inhibitor monotherapy can persist for months. In contrast, some patients do indeed clear FLT3-ITD from the marrow, although intervening marrow hypocellularity/aplasia is uncommon. During the Chrysalis trial, a highly sensitive NGS assay was used to serially quantify FLT3-ITD allele frequency in a subset of patients treated with doses ≥ 80 mg (n = 80) to determine whether FLT3-ITD burden was a biomarker for survival.41 This analysis showed that patients with a CRc and a ≥2-log reduction in FLT3-ITD compared with baseline experienced superior survival compared with patients with a comparable degree of morphologic response but without mutational clearance. Although a 4-log reduction in FLT3-ITD was observed in a subset of responders (11/44 studied patients with CRc), the depth of response did not predict the degree of peripheral count recovery, and OS was not obviously better than in patients with a ≥2-log reduction in FLT3-ITD from baseline.41 These preliminary data suggest that FLT3-ITD mutational clearance may be an important end point for future trials.
It is notable that, even when FLT3-ITD clears during gilteritinib response, restoration of normal nonclonal hematopoiesis is distinctly uncommon, except among patients who are subsequently transplanted or are treated for relapse after HSCT and reestablish full donor chimerism during gilteritinib response.83,99 For the remainder, the “recovering” marrow generally is clonal. Single-cell NGS analysis has actually confirmed polyclonality during gilteritinib response, which may include a dominant clone of differentiating FLT3-mutated cells or contraction of the FLT3 mutant clones coincident with expansion of FLT3 wild-type clones containing multiple leukemia-associated mutations (eg, NPM1, WT1, IDH1/2, TET2).83,99 These data paint a complicated picture of dynamic intratumoral heterogeneity under the selective pressures of FLT3-targeted therapy, with expansion of nontargeted clones, persistence of differentiating clones, and, ultimately, clonal evolution of either population to limit response durability in the absence of transplant.
Resistance to gilteritinib
Causes of primary resistance to gilteritinib are poorly understood. It has been hypothesized that prior midostaurin exposure could alter responsiveness to gilteritinib, but this has not clearly been seen anecdotally, and too few such patients enrolled in the Chrysalis or ADMIRAL trial to make any clear inferences. Causes of secondary drug resistance were recently described from 41 patients treated on gilteritinib monotherapy studies.99 New mutations were observed at disease progression and conferred in vitro drug resistance in FLT3-ITD AML cells. The most commonly observed clinical resistance mutations were NRAS, KRAS, and other MAPK pathway activating mutations (including rare BCR-ABL1 fusions). From single-cell NGS analysis, these resistance mutations indeed occurred in FLT3-mutated cells and, in some cases, preceded gilteritinib therapy.99 Additionally, a substantial number of patients lacked new mutations at progression or showed expansion of FLT3 wild-type clones with or without new mutations. On-target mutations in FLT3 were uncommon, exclusively occurred at F691L, and were only seen in patients treated at doses ≤120 mg.99 These data are consistent with preclinical observations that F691L mutation causes only a modest increase in IC50 in FLT3-ITD–mutated cells and argue for gilteritinib dose escalation if this mutation is experienced clinically.64
An obvious solution to avoid polyclonal drug-resistant R/R AML is to optimize therapy for newly diagnosed patients with FLT3-mutated AML. Frontline clinical trials combining gilteritinib with intensive induction chemotherapy or lower-toxicity agents have been initiated and show promising results.100 A multicenter phase 1b study conducted in newly diagnosed AML patients showed the tolerability of intensive induction plus full-dose gilteritinib (120 mg daily for 14 days per cycle starting after completion of anthracycline) added to full-dose cytarabine and anthracycline induction and high-dose cytarabine consolidation. In this trial, a very high CRc rate (89%) was seen among 27 newly diagnosed patients with FLT3 mutations. A trial adding gilteritinib to azacitidine in patients with newly diagnosed FLT3-mutated AML who were unfit for more intensive regimens has demonstrated the tolerability of this approach and showed a CRc rate of 10 of 15 from its safety run-in cohort.101 Long-term follow-up results are not yet available from either trial. However, based upon feasibility, activity, and expectation of potential benefit, randomized trials to compare these new approaches with current standards have been initiated worldwide, as have studies examining gilteritinib in the maintenance setting following standard chemotherapy or HSCT. Studies combining gilteritinib with other biologically targeted agents, including venetoclax, IDH inhibitors, and immunotherapeutics, are planned or already underway in the R/R setting; if results are promising, these combinations will certainly be moved earlier in therapy.
Conclusions and future directions
Gilteritinib stands as the remarkable product of a global translational science effort, and it has appropriately received regulatory approval for the treatment of R/R FLT3-mutated AML as monotherapy. However, as monotherapy, FLT3 inhibition, with gilteritinib or with any other potent FLT3 inhibitor,60 is unlikely to result in a high cure rate for this population. Gilteritinib will likely improve the cure rates for patients with FLT3-mutated AML, but only when incorporated into a broader treatment regimen, which could include chemotherapy, other targeted agents, and immunotherapy, including allogeneic transplant. A number of such clinical studies have already been launched. Not surprisingly, gilteritinib is synergistic with cytarabine and anthracyclines in cell line and murine models102 ; however, killing cell lines in vitro and making artificial tumors in mice shrink are relatively easy tasks. Incorporating this drug effectively into chemotherapy and transplant regimens in human patients is considerably more difficult and not just because of the usual problems with safety, tolerability, and drug interactions. For example, gilteritinib’s activity is likely to be blunted by FLT3 ligand, the levels of which increase precipitously following intensive chemotherapy. Infectious risks may be increased by the drug’s inhibitory effects on dendritic cells. The regulatory approval of gilteritinib for R/R FLT3-mutant AML patients is only the first step; there is still a great deal of work to be done before this drug’s full potential is reached.
Authorship
Contribution: M.L. and A.E.P. wrote the manuscript.
Conflict-of-interest disclosure: M.L. serves as a consultant for, and receives honoraria and research support from, Daiichi-Sankyo, Novartis, Astellas, Amgen, Arog, Menarini, and Agios. A.E.P. serves as a consultant for, and receives honoraria and research support from, Daiichi-Sankyo, Novartis, Astellas, Arog, AbbVie, and Agios.
Correspondence: Mark Levis, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, 1650 Orleans St, Room 2M44, Baltimore, MD 21287; e-mail: levisma@jhmi.edu.

