

Key Points
Co-inhibition of XPO1 and BCL2 induces apoptosis in AML cells and decreases leukemia burden in cell line and patient-derived xenografts.
Combining SINE compounds with venetoclax shrinks tumors and extends survival in DLBCL xenografts.

Abstract
The selective inhibitor of nuclear export (SINE) compounds selinexor (KPT-330) and eltanexor (KPT-8602) are from a novel class of small molecules that target exportin-1 (XPO1 [CRM1]), an essential nucleo-cytoplasmic transport protein responsible for the nuclear export of major tumor suppressor proteins and growth regulators such as p53, p21, and p27. XPO1 also affects the translation of messenger RNAs for critical oncogenes, including MYC, BCL2, MCL1, and BCL6, by blocking the export of the translation initiation factor eIF4E. Early trials with venetoclax (ABT-199), a potent, selective inhibitor of BCL2, have revealed responses across a variety of hematologic malignancies. However, many tumors are not responsive to venetoclax. We used models of acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL) to determine in vitro and in vivo responses to treatment with venetoclax and SINE compounds combined. Cotreatment with venetoclax and SINE compounds demonstrated loss of viability in multiple cell lines. Further in vitro analyses showed that this enhanced cell death was the result of an increase in apoptosis that led to a loss of clonogenicity in methylcellulose assays, coinciding with activation of p53 and loss of MCL1. Treatment with SINE compounds and venetoclax combined led to a reduction in tumor growth in both AML and DLBCL xenografts. Immunohistochemical analysis of tissue sections revealed that the reduction in tumor cells was partly the result of an induction of apoptosis. The enhanced effects of this combination were validated in primary AML and DLBCL patient cells. Our studies reveal synergy with SINE compounds and venetoclax in aggressive hematologic malignancies and provide a rationale for pursuing this approach in a clinical trial.
Introduction
Exportin-1 (XPO1 [CRM1]) is a well characterized and essential nucleo-cytoplasmic transport protein in the karyopherin family and is responsible for the nuclear export of nearly all major tumor suppressor proteins (TSPs), including p53, p21, FOXO, and IκB, regardless of their heterogeneous structures.1-4 Because TSPs depend on nuclear localization to perform genomic maintenance, many cancer cells increase the nuclear export of TSPs to inactivate critical DNA surveillance.5 In addition, XPO1 is responsible for exporting the translation initiation factor eIF4E (through the chaperone protein LRPPRC), which binds the capped messenger RNAs (mRNAs) of notable oncoproteins such as BCL2, MYC, and BCL6 and transports them to the cytoplasm for their translation.6 Given the key role of XPO1 in regulating these critical proteins, it is not surprising that XPO1 overexpression in numerous solid and hematologic malignancies often correlates with poor prognosis.2-5,7
Selective inhibitor of nuclear export (SINE) compounds and XPO1 antagonists are included in a novel class of small molecules that demonstrate therapeutic potential across a broad range of diseases, including inflammatory diseases, neurodegenerative disorders, and both solid and hematologic malignancies.1,7-12 Selinexor (KPT-330) is a first-in-class oral antagonist of XPO1 that inactivates XPO1 function by forming a slowly reversible covalent bond. In vitro studies have demonstrated cell cycle arrest, accumulation of DNA damage, and induction of apoptosis in response to treatment with SINE compounds as a result of the nuclear accumulation and reactivation of TSPs and reduced oncoproteins.1,2,13-15 We and others have previously shown that selinexor has anti-tumor activity in preclinical models of acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), T-cell acute lymphoblastic leukemia, multiple myeloma, and diffuse large B-cell lymphoma (DLBCL).2-4,15-19 In addition, selinexor is being tested as a single agent and in combination in ongoing phase 1 to 3 clinical trials for the treatment of advanced solid tumor and hematologic malignancies.7,10-12,20-22 A second-generation SINE compound, eltanexor (KPT-8602), has pharmacokinetic properties similar to those of selinexor, but it has decreased brain penetration, which allows for more frequent dosing and better tolerability and efficacy across preclinical models.23-25 Selinexor and eltanexor have been shown to specifically target cancer cells in both in vitro and in vivo tests.7,10-12,16,20-23,25 Reversible nausea, fatigue, anorexia, and thrombocytopenia seen in the clinic can be mitigated with standard antiemetic regimens and dose reductions.26 These toxicities are dose-dependent, and thus, dose-sparing combination strategies that maximize anti-tumor activity while minimizing toxicity are needed.
Apoptosis is controlled by the BCL2 family of proteins within the mitochondria. Given that evasion of apoptosis is a hallmark of cancer, it is not surprising that anti-apoptotic proteins BCL2 and MCL1 are dysregulated and linked to maintenance and survival in numerous malignancies.27-29 These proteins inhibit apoptosis through the sequestration of the pro-apoptotic protein BIM, which is required for activation of BAX/BAK and the subsequent induction of mitochondrial outer membrane permeabilization.27,28 Earlier, these proteins were a challenge to target with small molecules, but clinically relevant inhibitors have recently been synthesized.30-33 Early studies with venetoclax (ABT-199), a potent and selective inhibitor of BCL2, have revealed impressive response rates across a variety of hematologic malignancies.30,34-39 Responses are nearly universal in CLL, a malignancy almost entirely reliant on BCL2.36 However, in some aggressive hematologic malignancies, such as AML and DLBCL, anti-apoptotic dependence is more heterogeneous with MCL1-, BCL-xL–, and BCL2-dependent clones, which are often present within a single patient’s tumor. Thus, in these malignancies, treatment with venetoclax may select for upregulation of other BCL2 family anti-apoptotic proteins, most commonly MCL1, and switching of anti-apoptotic dependence.40-43 It has been suggested that the simultaneous inhibition of BCL2 and MCL1 may be required for a more durable response.32,33,44 Interestingly, the mRNAs for BCL2 and MCL1 are both controlled by the XPO1-regulated protein eIF4E through either direct transportation from the nucleus to the cytoplasm for translation (through the chaperone protein LRPPRC) or by enhanced translation of their complicated 5′ untranslated region mRNA structures.6,45-47 Because XPO1 is inactivated by SINE compounds, treatment with venetoclax and SINE compounds combined has the potential to be more effective against these malignancies than venetoclax alone. Here, we show that cotreatment with venetoclax and SINE compounds results in enhanced cell killing in both in vitro and in vivo models of aggressive hematologic malignancies.
Methods
Patient samples
Experiments were conducted on primary AML or DLBCL patient samples with a baseline viability of >60%, which were provided by the Vanderbilt-Ingram Cancer Center Hematologic Malignancy Tumor Bank in accordance with the tenets of the Declaration of Helsinki. The study was approved by the Vanderbilt University Medical Center Institutional Review Board. Normal CD34+ cells were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA).
Drug combination analysis and calculation of synergy
To assess the efficacy of the combination of venetoclax and SINE compounds, cells were plated, and viability was determined as described above by using twofold to threefold dilution matrices of SINE and venetoclax. The effects of the combinations were calculated by using the zero interaction potency (ZIP) model, which compares observed and expected combination effects.48 The ZIP model combines the advantages of the Bliss independence and Loewe additivity models. As a null hypothesis, it assumes that 2 noninteracting drugs will incur minimal changes in their drug response curve. The output of the ZIP model is the delta synergy score (δ), which is the average combination effect of the drugs over the total dose matrix tested. A δ score of 0 implies both probabilistic independence and dose additivity, whereas a δ score of <0 indicates antagonistic drug effects and a score of >0 indicates synergistic drug effects.
MV-4-11 and patient-derived AML xenograft mouse models
MV-4-11 or patient bone marrow cells were intravenously injected into the tail vein of female NSGS mice 1 day after they received low-dose irradiation (1 Gy). For the MV-4-11 model, the leukemia was allowed to engraft for 6 days, and mice were randomly assigned to oral gavage with vehicle control, eltanexor (7.5 mg/kg), venetoclax (25 mg/kg), or the combination of eltanexor (7.5 mg/kg) and venetoclax (25 mg/kg) for 5 days per week for 3 weeks. For the patient-derived xenograft (PDX) model, mice were randomly assigned to groups with 5 mice per group for equal distribution of peripheral blood chimerism 7 weeks after injection. Mice were treated by oral gavage starting at week 8 with vehicle control, eltanexor (7.5 mg/kg), venetoclax (25 mg/kg), or a combination of eltanexor (7.5 mg/kg) and venetoclax (25 mg/kg) for 5 days per week for 6 weeks. For all mice that received a transplantation, blood was drawn once per week to monitor the development of leukemia, and the mice were closely monitored for signs of leukemia progression such as weight loss, lethargy, and hind limb paralysis.
DoHH-2 xenograft mouse model
Cultured DoHH-2 cells were harvested during log phase growth and resuspended in cold phosphate-buffered saline at a concentration of 1 × 108 cells per mL. Xenografts were initiated by subcutaneously implanting 1 × 107 DoHH-2 cells (0.1 mL suspension) into the right flank of each test animal, and tumors were monitored as their volumes approached the target range of 100 to 150 mm3. For the eltanexor studies (Figure 4A-C), mice were randomly sorted into 4 efficacy groups (with 8 mice per group) 13 days after implantation (designated as day 1 of the study). The group mean tumor volume was ∼120 mm3. Mice were dosed with vehicle once per day for 5 days, 10 mg/kg eltanexor once per day for 5 days (dose reduced to 7.5 mg/kg on day 9 because of presumed treatment-related weight loss), 100 mg/kg venetoclax once per day, or a combination of 10 mg/kg eltanexor once per day for 5 days (reduced to 7.5 mg/kg on day 9) plus 100 mg/kg venetoclax once per day. Similarly, for the selinexor studies (supplemental Figure 7C-E), mice were randomly sorted into 4 efficacy groups (8 mice per group) 10 days after implantation (designated as day 1 of the study). The group mean tumor volume was ∼115 mm3. Mice were dosed with vehicle once per day for 5 days, 10 mg/kg selinexor every other day for 3 days, 100 mg/kg venetoclax once per day, or a combination of 10 mg/kg selinexor every other day for 3 days plus 100 mg/kg venetoclax once per day.
Toledo xenograft mouse model
Toledo cells were harvested at log growth phase and resuspended in phosphate-buffered saline at 5 × 107 cells per mL. Cells were placed on ice and mixed with an equal volume of Matrigel (Corning 354234). The mixture was kept on ice and injected into the left flank of mice in a volume of 0.2 mL (equivalent to 5 × 106 cells per mouse). Forty female NCr nude mice (Taconic) were inoculated subcutaneously in the left flank with 1 × 107 Toledo cells. Treatment was initiated when the tumors reached a mean volume of 802.7 mm3. Mice were randomly allocated into 4 groups of 10 mice each such that mean tumor volume in each group was ∼800 mm3. Mice were treated with vehicle every other day for 3 days, 5 mg/kg selinexor every other day for 3 days, 25 mg/kg venetoclax once per day, or a combination of 5 mg/kg selinexor every other day for 3 days plus 25 mg/kg venetoclax once per day.
Results
Leukemia cell lines are differentially sensitive to XPO1 and BCL2 inhibition in vitro
To determine cell specificity to XPO1 inhibition, we treated a panel of leukemia cell lines with selinexor and eltanexor. Cell viability was measured after treatment using the CellTiter-Glo viability assay, and the 50% growth inhibition concentration (GI50) was calculated (Table 1; supplemental Figure 1A). Congruent with previous findings, selinexor and eltanexor had broad activity across multiple cell lines.23 To confirm on-target drug activity, we performed a dose titration of eltanexor in MV-4-11 and MOLM-13 cells and saw a dose-dependent reduction in XPO1 protein levels (Figure 1). We also assessed the effect of eltanexor on other cargo proteins known to be regulated by XPO1 and discovered a dose-dependent increase in p53 with a concurrent decrease in MCL1 levels. These findings coincided with increasing levels of cleaved caspase 3 (Figure 1). The recent finding that pharmacologic activation of p53 is synergistic with BCL2 inhibition underscored our expectation for enhanced cellular death by combining SINE compounds with venetoclax.49 Although nearly all cell lines tested showed decreased viability upon inhibition of XPO1, as expected, there was a clear divide in the cells in terms of their sensitivity to BCL2 inhibition linked to the functional p53 status of the cells.49-52 This was confirmed by treatment with the MDM2 inhibitor RG7388. Although p53wild-type MOLM-13 and MV-4-11 cells were sensitive to venetoclax treatment, p53mutant NB4 cells were mildly resistant, and p53mutant MOLM-16 and p53null K-562 and U-937 cells were completely resistant (Table 1; supplemental Figure 1A-B).
SINE compounds and venetoclax display differential growth inhibition of malignant myeloid cell lines
| Cell line | Average GI50 at 72 h, nM | ||
|---|---|---|---|
| Selinexor | Eltanexor | Venetoclax | |
| MOLM-13 | 52 | 32 | 25 |
| MV-4-11 | 57 | 22 | 52 |
| K-562 | 241 | 104 | >5000 |
| MOLM-16 | 165 | 59 | >5000 |
| U-937 | 444 | 131 | >5000 |
| NB4 | 571 | 264 | >1000 |
| Cell line | Average GI50 at 72 h, nM | ||
|---|---|---|---|
| Selinexor | Eltanexor | Venetoclax | |
| MOLM-13 | 52 | 32 | 25 |
| MV-4-11 | 57 | 22 | 52 |
| K-562 | 241 | 104 | >5000 |
| MOLM-16 | 165 | 59 | >5000 |
| U-937 | 444 | 131 | >5000 |
| NB4 | 571 | 264 | >1000 |
Values are averaged from 3 independent experiments.
![Treatment with eltanexor leads to alterations in apoptotic proteins, and combination with venetoclax synergizes to inhibit growth of AML cells in vitro. Western blot analysis of MV-4-11 and MOLM-13 cells after 24 hours of treatment with dimethyl sulfoxide (DMSO [D]) or a dose titration of eltanexor (ELTA) to determine protein levels of XPO1, p53, MCL1, and cleaved caspase 3. Actin was used as a loading control. Vertical line indicates different exposures because of varying levels of endogenous XPO1.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/3/10.1182_bloodadvances.2019000359/2/m_advancesadv2019000359f1.png?Expires=1767697652&Signature=AknpBnvXiCJ6K6sr7zOmjPKYgdXb7cD0j1oNNHd6-T2L1S~xhhG7Cq0F-Pi~1Aho~RZ-1HKY5qwF8s9UHYhE0fY09XfXpLfSSxEPHs9ejw9vWW1otZn6dvASLMhOsKbH0AES9jOqamr-e4UnXaF9LJ-2CX4LvsDNnRX8z7kufxlsTXGY3hqx4-rfW5IvOJv9GV4SzxHzubk4luURGy3gUQ2n975-ux1vQXh3RkV9te2niymJWf2cKxh9C5kwJliHCDpd9zJW6WS-Idblx8ixfm-kZatsX1KZiT2iSu4G2pcuXng6ezJmZjms--BUijwavhhIyhi9i1PUyx4RCbNwUA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Treatment with eltanexor leads to alterations in apoptotic proteins, and combination with venetoclax synergizes to inhibit growth of AML cells in vitro. Western blot analysis of MV-4-11 and MOLM-13 cells after 24 hours of treatment with dimethyl sulfoxide (DMSO [D]) or a dose titration of eltanexor (ELTA) to determine protein levels of XPO1, p53, MCL1, and cleaved caspase 3. Actin was used as a loading control. Vertical line indicates different exposures because of varying levels of endogenous XPO1.
Treatment with eltanexor leads to alterations in apoptotic proteins, and combination with venetoclax synergizes to inhibit growth of AML cells in vitro. Western blot analysis of MV-4-11 and MOLM-13 cells after 24 hours of treatment with dimethyl sulfoxide (DMSO [D]) or a dose titration of eltanexor (ELTA) to determine protein levels of XPO1, p53, MCL1, and cleaved caspase 3. Actin was used as a loading control. Vertical line indicates different exposures because of varying levels of endogenous XPO1.
SINE compounds and venetoclax synergize in AML cells to reduce cell viability and increase apoptosis leading to loss of clonogenicity
To test whether co-inhibition of XPO1 and BCL2 would be synergistic in AML, we performed combination studies with twofold dilutions of eltanexor and venetoclax. After 72 hours of exposure, cell viability was measured using the CellTiter-Glo assay. As might be expected, there was no additional effect from adding venetoclax to eltanexor in the venetoclax-resistant cells (MOLM-16, K-562, and U-937). However, this combined treatment resulted in ZIP δ synergy scores >0 in the MOLM-13, MV-4-11, and NB4 cells (Figure 2A; supplemental Figure 1C-D), suggesting a synergistic effect on cell viability in these cell lines. Consistent with these results, cell counts with trypan blue exclusion showed a reduction in the number of viable MV-4-11, MOLM-13, and NB4 cells in the combination treatment group (supplemental Figure 2). Similar results were found when venetoclax was combined with selinexor in MOLM-13 and MV-4-11 cells (NB4 cells were not tested with the selinexor combination) (supplemental Figure 3).

Combination of eltanexor and venetoclax induces apoptosis and leads to loss of clonogenicity. (A) MV-4-11, MOLM-13, and NB4 cells were treated with twofold dilutions of eltanexor, venetoclax, or their combination for 72 hours. Contour plots of synergy scores were generated from a dose matrix of eltanexor and venetoclax using the ZIP model. The synergy scores were represented by pseudocoloring 2-dimensional (left) or 3-dimensional (right) contour plots over the dose matrix, giving rise to the overall synergy landscape. Red indicates synergy and green indicates antagonism for the various concentrations of combined eltanexor and venetoclax. Note different scale bars for ZIP synergy scores for the cell lines. Plots are representative of 3 independent experiments. (B) p53, BCL2, MCL1, and PARP and cleaved PARP (PARP/cPARP) protein expression was measured by immunoblot in MV-4-11, MOLM-13, and NB4 cells after 24-hour treatment with DMSO (D), eltanexor (E), venetoclax (V), or eltanexor-venetoclax combined (E/V). Actin was used as a loading control. Vertical lines indicate different exposures because of varying levels of endogenous protein levels between the cell lines. (C) Apoptosis was measured by annexin V (AnnV)/PI staining using flow cytometry after 24 hours of treatment. The percentages of early apoptotic (AnnV+; blue bar) and late apoptotic/dead (AnnV+/PI+; red bar) cells were measured. Statistical differences were calculated on the basis of the total number of AnnV+ cells for each treatment group. (D) Colony assays in MV-4-11, MOLM-13, and NB4 cells after 24 hours of drug treatment. The plot shows number of colonies per 500 cells plated. The data in panels C-D are presented as mean ± standard deviation (SD) from 3 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Combination of eltanexor and venetoclax induces apoptosis and leads to loss of clonogenicity. (A) MV-4-11, MOLM-13, and NB4 cells were treated with twofold dilutions of eltanexor, venetoclax, or their combination for 72 hours. Contour plots of synergy scores were generated from a dose matrix of eltanexor and venetoclax using the ZIP model. The synergy scores were represented by pseudocoloring 2-dimensional (left) or 3-dimensional (right) contour plots over the dose matrix, giving rise to the overall synergy landscape. Red indicates synergy and green indicates antagonism for the various concentrations of combined eltanexor and venetoclax. Note different scale bars for ZIP synergy scores for the cell lines. Plots are representative of 3 independent experiments. (B) p53, BCL2, MCL1, and PARP and cleaved PARP (PARP/cPARP) protein expression was measured by immunoblot in MV-4-11, MOLM-13, and NB4 cells after 24-hour treatment with DMSO (D), eltanexor (E), venetoclax (V), or eltanexor-venetoclax combined (E/V). Actin was used as a loading control. Vertical lines indicate different exposures because of varying levels of endogenous protein levels between the cell lines. (C) Apoptosis was measured by annexin V (AnnV)/PI staining using flow cytometry after 24 hours of treatment. The percentages of early apoptotic (AnnV+; blue bar) and late apoptotic/dead (AnnV+/PI+; red bar) cells were measured. Statistical differences were calculated on the basis of the total number of AnnV+ cells for each treatment group. (D) Colony assays in MV-4-11, MOLM-13, and NB4 cells after 24 hours of drug treatment. The plot shows number of colonies per 500 cells plated. The data in panels C-D are presented as mean ± standard deviation (SD) from 3 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
We performed western blot analysis to determine key protein changes that would cause a loss of viability. First, we measured steady-state p53 protein levels because p53 is a known XPO1-regulated tumor suppressor protein that affects apoptosis. Although p53 in NB4 cells (nonfunctional mutant p53) was not affected, treatment with eltanexor led to an increase in p53 in both cell lines with functional p53: MOLM-13, and MV-4-11. In addition, eltanexor decreased MCL1 in all 3 cell lines; reduction of MCL1 was enhanced with the combination treatment. Concordant with increased p53 and reduction in MCL1, eltanexor increased cleaved poly (ADP-ribose) polymerase (PARP) in all cell lines tested, indicating active engagement in apoptosis. Moreover, the loss of cell proliferation seen with the eltanexor-venetoclax combination coincided with further increase in cleaved PARP in the combination groups. There were no changes in BCL2 protein levels with either treatment, consistent with previous independent studies of both SINE compounds and venetoclax (Figure 2B).30,53 Lentiviral knockdown of TP53 in MOLM-13 and MV-4-11 cells displayed minimal reduction in apoptotic cells after treatment with eltanexor and venetoclax, but the loss of p53 had minimal impact on the caspase-dependent loss of cell viability induced by the combination treatment (supplemental Figure 4).
As further evidence that eltanexor and venetoclax lead to apoptosis, cells were stained with annexin V (annV) and propidium iodine (PI), and the cells treated with the eltanexor and venetoclax combination displayed an enhanced induction of apoptosis (annV+) and cell death (annV+/PI+) compared with single treatment with eltanexor or venetoclax (Figure 2C; supplemental Figure 5). To determine the long-term effects that reduction in proliferation and increased apoptosis would have on the leukemic potential of the cells, colony-forming unit assays were performed on MOLM-13, MV-4-11, and NB4 cells after 24 hours of treatment with eltanexor, venetoclax, or the combination of both. Although treatment with eltanexor or venetoclax alone led to a reduction of 10% to 40% in colonies, treatment with eltanexor-venetoclax combined led to a greater than 60% to 90% reduction in colonies, indicating an enhanced loss of clonogenicity of the leukemia cells (Figure 2D).
Combination of eltanexor and venetoclax enhances their anti-leukemic effects in murine AML cell lines and PDX models
To examine the in vivo efficacy of treatment with SINE compounds plus venetoclax, we evaluated an MV-4-11 xenograft mouse model of AML. MV-4-11 cells were injected into the tail vein of NSGS mice. Starting 1 week after injection, the mice were given vehicle control, eltanexor (7.5 mg/kg), venetoclax (25 mg/kg), or eltanexor-venetoclax combined 5 days a week for 3 weeks. Once vehicle treated mice began to succumb to leukemia, all of the treatment groups were euthanized to determine the amount of leukemia cells in all of the hematopoietic tissues. Upon gross examination, it was apparent that all treatments decreased the spleen weight compared with vehicle control without affecting total body weight (supplemental Figure 6A-B). After isolating cells from the peripheral blood, bone marrow, and spleen, flow cytometry analysis for human CD45+ cells revealed a significant reduction in the leukemia burden in all 3 compartments analyzed after treatment with eltanexor and venetoclax as single agents. Treatment with eltanexor-venetoclax combined significantly diminished the leukemia burden beyond what was achieved by treatment with eltanexor and venetoclax as single agents in all hematopoietic tissues and nearly ablated leukemia cells from the peripheral blood and spleen (Figure 3A-B; supplemental Figure 6C). Hematoxylin and eosin staining of bone marrow and spleen sections from treated mice exemplified the decrease of human AML cells in both tissues, particularly in the group of mice treated with eltanexor-venetoclax combined, which displayed some restoration of normal mouse hematopoietic cellular architecture in both of these hematopoietic compartments (Figure 3C; supplemental Figure 6D).
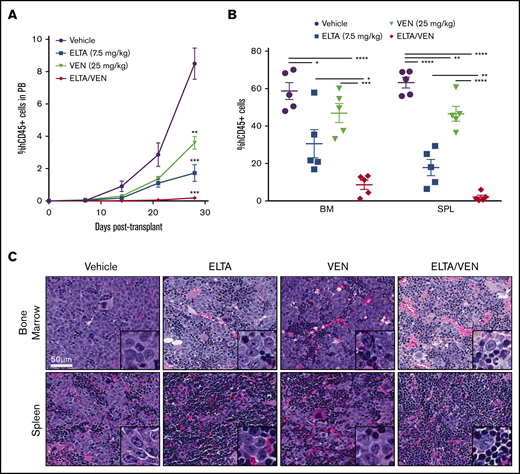
Combination of eltanexor and venetoclax eliminates leukemia cells in an MV-4-11 AML xenograft model. An MV-4-11 model was used to test the in vivo effects of eltanexor (ELTA), venetoclax (VEN), and their combination (ELTA/VEN) treatment compared with vehicle. (A) Percentage of human CD45+ (hCD45+) cells in the peripheral blood (PB) from day 7 to day 28 posttransplantation. **P < .01 and ***P < .001, compared with vehicle at day 28. (B) Percentage of hCD45+ cells in the bone marrow (BM) and spleen (SPL) tissues at day 28 posttransplantation. (C) Hematoxylin and eosin staining of bone marrow and spleen from the mice in panels A and B treated with vehicle, eltanexor, venetoclax, or eltanexor-venetoclax combined. Scale bar, 50 µm. Histology images were acquired with an Aperio AT Turbo scanner at an original magnification of ×40; inset box, original magnification, ×100. The data represent the mean of 5 mice per cohort ± SD. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Combination of eltanexor and venetoclax eliminates leukemia cells in an MV-4-11 AML xenograft model. An MV-4-11 model was used to test the in vivo effects of eltanexor (ELTA), venetoclax (VEN), and their combination (ELTA/VEN) treatment compared with vehicle. (A) Percentage of human CD45+ (hCD45+) cells in the peripheral blood (PB) from day 7 to day 28 posttransplantation. **P < .01 and ***P < .001, compared with vehicle at day 28. (B) Percentage of hCD45+ cells in the bone marrow (BM) and spleen (SPL) tissues at day 28 posttransplantation. (C) Hematoxylin and eosin staining of bone marrow and spleen from the mice in panels A and B treated with vehicle, eltanexor, venetoclax, or eltanexor-venetoclax combined. Scale bar, 50 µm. Histology images were acquired with an Aperio AT Turbo scanner at an original magnification of ×40; inset box, original magnification, ×100. The data represent the mean of 5 mice per cohort ± SD. *P < .05; **P < .01; ***P < .001; ****P < .0001.
SINE-venetoclax combination has synergistic anti-tumor activity and prolongs survival in DLBCL xenograft models
DLBCL is an aggressive form of non-Hodgkin lymphoma, and 90% of relapsed or refractory patients are currently incurable, which indicates a need for better treatment options for this subset of patients.54 One of the most common mechanisms of action that leads to the poor prognosis in DLBCL is overexpression of BCL2 or MCL1 for evading apoptosis.42,54,55 Treatment of DLBCL cells with venetoclax as a single agent yields mixed results, and similar to AML cells, is accompanied by upregulation of MCL1.42,54
To test whether the eltanexor-venetoclax combination would synergize in DLBCL, SCID mice had DoHH-2 (double-hit DLBCL; MYC and BCL2 rearrangement) cells subcutaneously implanted in their hind quarters. The tumors grew to ∼100 mm3 mean size and mice with these tumors were randomly assigned to 1 of 4 treatment groups: vehicle control, eltanexor (10 mg/kg), venetoclax (100 mg/kg), or eltanexor-venetoclax combined. The eltanexor dose was reduced on day 9 during the study (7.5 mg/kg) because of weight loss in the mice, which was arrested with the new dose (supplemental Figure 7A). There was a clear treatment benefit with each of the single agents when compared with vehicle control. When mice were treated with eltanexor-venetoclax combined, they demonstrated tumor regression at certain time points with an overall benefit when compared with mice treated with vehicle or single agents. On day 14 of treatment, DoHH-2 tumor volumes had tumor growth inhibition of 53% for eltanexor, 59% for venetoclax, and 97% for eltanexor-venetoclax combined when compared with vehicle control. (Figure 4A; supplemental Figure 7B). Furthermore, this combination treatment resulted in a distinguishable survival benefit when compared with vehicle (Figure 4B).
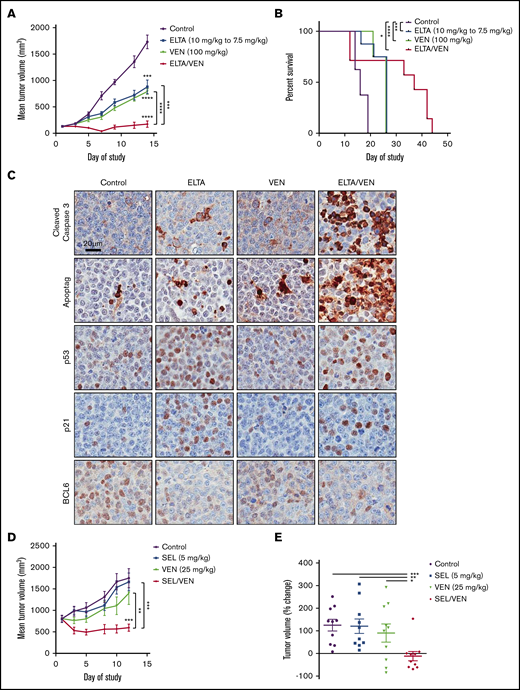
SINE-venetoclax combination inhibits tumor growth and prolongs survival in DLBCL mouse xenograft models. (A) A DoHH-2 xenograft model was used to test the in vivo efficacy of treatment with eltanexor, venetoclax, or eltanexor-venetoclax combined compared with vehicle control. The data represent the mean of 6 to 8 mice per cohort ± standard error of the mean (SEM). (B) Kaplan-Meier survival of mouse cohorts (n = 7-8 mice each), indicating median survival for mice treated with vehicle (16 days), eltanexor (26 days), venetoclax (26 days), or eltanexor-venetoclax combined (37 days). (C) Representative immunohistochemical staining of tumor sections for expression of cleaved caspase 3, ApopTag, p53, p21, and BCL6 from DoHH-2 xenograft mice in panel A. Scale bar, 20 µm. Histology images were acquired with an Aperio AT Turbo scanner at 20× magnification. (D) A Toledo xenograft model was allowed to grow to an average volume of 800 mm3 before testing the in vivo efficacy of treatment with selinexor, venetoclax, or selinexor-venetoclax combined compared with vehicle. The data represent the mean of 10 mice per cohort ± SEM. (E) The percentage change of the vehicle and 3 treatment groups on day 12 was normalized to the baseline measurement of each group on day 1 of treatment. The data represent the mean of 10 mice per cohort ± SEM. *P < .05; **P < .01; ***P < .001; ****P < .0001.
SINE-venetoclax combination inhibits tumor growth and prolongs survival in DLBCL mouse xenograft models. (A) A DoHH-2 xenograft model was used to test the in vivo efficacy of treatment with eltanexor, venetoclax, or eltanexor-venetoclax combined compared with vehicle control. The data represent the mean of 6 to 8 mice per cohort ± standard error of the mean (SEM). (B) Kaplan-Meier survival of mouse cohorts (n = 7-8 mice each), indicating median survival for mice treated with vehicle (16 days), eltanexor (26 days), venetoclax (26 days), or eltanexor-venetoclax combined (37 days). (C) Representative immunohistochemical staining of tumor sections for expression of cleaved caspase 3, ApopTag, p53, p21, and BCL6 from DoHH-2 xenograft mice in panel A. Scale bar, 20 µm. Histology images were acquired with an Aperio AT Turbo scanner at 20× magnification. (D) A Toledo xenograft model was allowed to grow to an average volume of 800 mm3 before testing the in vivo efficacy of treatment with selinexor, venetoclax, or selinexor-venetoclax combined compared with vehicle. The data represent the mean of 10 mice per cohort ± SEM. (E) The percentage change of the vehicle and 3 treatment groups on day 12 was normalized to the baseline measurement of each group on day 1 of treatment. The data represent the mean of 10 mice per cohort ± SEM. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Immunohistochemistry was used to determine the effects of eltanexor and venetoclax, alone or in combination, on the induction of apoptosis and the expression of select TSPs and oncoproteins in DoHH-2–derived DLBCL xenograft models. Compared with samples treated with vehicle control, samples treated with eltanexor and venetoclax as single agents had significantly increased apoptosis, as shown by cleaved caspase 3 and ApopTag staining. Apoptosis was further increased in samples treated with eltanexor-venetoclax combined. Increased nuclear staining of p53 and p21 was observed in samples treated with single-agent eltanexor and with eltanexor-venetoclax combined. BCL6 nuclear staining was downregulated in samples treated with eltanexor alone or eltanexor-venetoclax combined (Figure 4C). Similar results were seen in a separate DoHH-2 xenograft study in which mice were treated with vehicle, selinexor (10 mg/kg), venetoclax (100 mg/kg), or selinexor-venetoclax combined (supplemental Figure 7C-E).
To test the ability of the SINE compound plus venetoclax combination to affect larger tumors, the Toledo cell line was implanted subcutaneously in the hind quarter of NCr mice. For this study, the tumors were allowed to grow to a mean size of ∼800 mm3; the mice were then randomly split into 4 treatment groups: vehicle control, selinexor (5 mg/kg), venetoclax (25 mg/kg), or selinexor-venetoclax combined. In this case, neither single agent significantly impeded the rapid growth of these large tumors, but treatment with selinexor-venetoclax combined led to the complete inhibition of tumor growth and even a slight regression of tumors without affecting total body weight (Figure 4D-E; supplemental Figure 7F-G). The percentage change of tumor growth from day 0 to day 12 was 125% for vehicle, 121% for selinexor, 90% for venetoclax, and −12% for selinexor-venetoclax combined (Figure 4E; supplemental Figure 7G).
SINE and venetoclax synergize to reduce viability of AML and DLBCL patient cells ex vivo
To determine the effects of treatment with combined SINE and venetoclax on primary human cells (supplemental Table 1), we treated AML and DLBCL bone marrow mononuclear cells with eltanexor and venetoclax for 48 hours before measuring cell viability using the CellTiter-Glo assay. Treatment with eltanexor and venetoclax significantly reduced cell viability in the primary DLBCL cells compared with treatment with venetoclax alone (Figure 5A) and significantly reduced cell viability in the primary AML cells compared with treatment using eltanexor and venetoclax separately (Figure 5B) with corresponding reductions in GI50 for both patient populations (supplemental Figures 8D and 9B). Concordantly, staining with annV and flow cytometry revealed a significant reduction in the percent of live AML blasts (SSClo/CD45mid/CD33+) after treatment with eltanexor and venetoclax when compared with treatment using venetoclax alone with a smaller loss of normal myeloid blasts (Figure 5C). In addition, the combined treatment resulted in ZIP δ synergy scores >0 in all of the 13 AML samples tested, and 3 of 4 DLBCL samples tested (Table 2; supplemental Figures 8 and 9), which suggests an overall synergistic effect on cell viability in the primary AML and DLBCL cells. Moreover, treatment with both SINE and venetoclax was synergistic in samples from AML patients who responded to venetoclax plus DNA methyltransferase inhibiton in the clinic or patients who had primary induction failure to venetoclax in the clinic, regardless of p53 status (Figure 5B-C; Table 2, patients AML-009 to AML-013; supplemental Figure 8B-C).
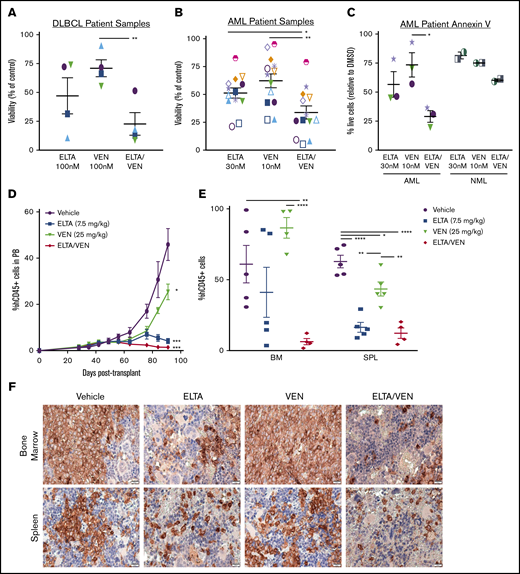
Synergistic anti-tumor effects of eltanexor and venetoclax in primary AML and DLBCL samples ex vivo and in an AML patient-derived xenograft. AML and DLBCL mononuclear cells were treated ex vivo with threefold dilutions of eltanexor, venetoclax, or eltanexor-venetoclax combined for 48 hours. The viability of treated cells compared with DMSO control using the CellTiter-Glo assay was plotted for the indicated concentrations. Viability was significantly decreased after 48 hours of treatment with combined eltanexor-venetoclax in DLBCL (A) or AML (B) cells. (C) Apoptosis was measured in AML or normal (NML) blast cells (SSClo/CD45mid/CD33+) by AnnV/PI staining using flow cytometry after 24 hours of treatment. The percent of live cells (AnnV–/PI–) relative to DMSO was measured. Data are expressed as mean ± SEM. Individual patients are represented by specific shapes in panels A-C, and individual AML patients who were venetoclax-responsive (orange) or primary induction refractory (lavender) in the clinic are represented by colored symbols in panels B and C. (D-E) An AML patient-derived xenograft model was used to test the in vivo effects of treatment with eltanexor, venetoclax, or eltanexor-venetoclax combined compared with vehicle. (D) Percentage of hCD45+ cells in the PB from day 28 to day 93 posttransplantation. The data represent the mean of 5 mice per cohort ± SEM. *P < .05 and ***P < .001, compared with vehicle at day 93. (E) Percentage of hCD45+ cells in the bone marrow and spleen tissues at day 93 posttransplantation. The data represent the mean of 5 mice per cohort ± SEM. (F) Representative immunohistochemical staining of bone marrow and spleen sections for expression of hCD45 from mice with AML patient-derived xenografts in panels D and E. Scale bar, 20 µm. Histology images were acquired with an Olympus BX43F microscrope at 40× magnification. *P < .05; **P < .01; ****P < .0001.
Synergistic anti-tumor effects of eltanexor and venetoclax in primary AML and DLBCL samples ex vivo and in an AML patient-derived xenograft. AML and DLBCL mononuclear cells were treated ex vivo with threefold dilutions of eltanexor, venetoclax, or eltanexor-venetoclax combined for 48 hours. The viability of treated cells compared with DMSO control using the CellTiter-Glo assay was plotted for the indicated concentrations. Viability was significantly decreased after 48 hours of treatment with combined eltanexor-venetoclax in DLBCL (A) or AML (B) cells. (C) Apoptosis was measured in AML or normal (NML) blast cells (SSClo/CD45mid/CD33+) by AnnV/PI staining using flow cytometry after 24 hours of treatment. The percent of live cells (AnnV–/PI–) relative to DMSO was measured. Data are expressed as mean ± SEM. Individual patients are represented by specific shapes in panels A-C, and individual AML patients who were venetoclax-responsive (orange) or primary induction refractory (lavender) in the clinic are represented by colored symbols in panels B and C. (D-E) An AML patient-derived xenograft model was used to test the in vivo effects of treatment with eltanexor, venetoclax, or eltanexor-venetoclax combined compared with vehicle. (D) Percentage of hCD45+ cells in the PB from day 28 to day 93 posttransplantation. The data represent the mean of 5 mice per cohort ± SEM. *P < .05 and ***P < .001, compared with vehicle at day 93. (E) Percentage of hCD45+ cells in the bone marrow and spleen tissues at day 93 posttransplantation. The data represent the mean of 5 mice per cohort ± SEM. (F) Representative immunohistochemical staining of bone marrow and spleen sections for expression of hCD45 from mice with AML patient-derived xenografts in panels D and E. Scale bar, 20 µm. Histology images were acquired with an Olympus BX43F microscrope at 40× magnification. *P < .05; **P < .01; ****P < .0001.
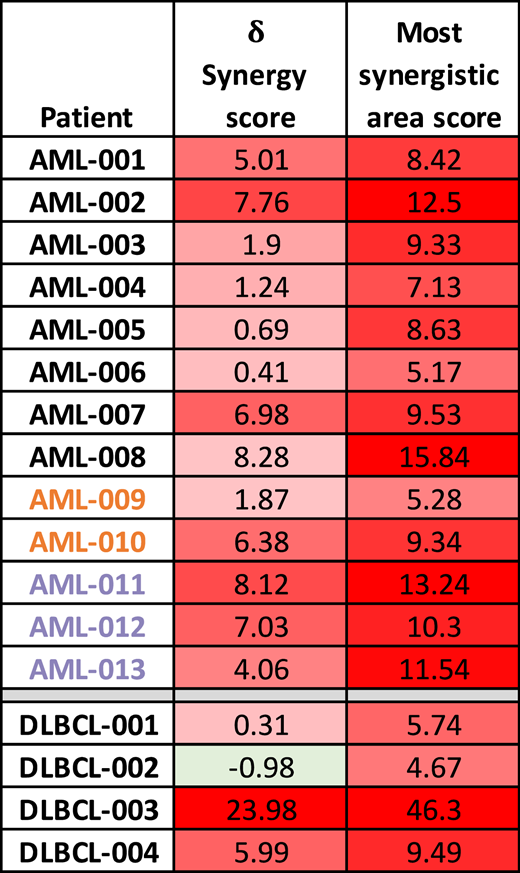
Overall ZIP δ synergy score and most synergistic area score for each AML and DLBCL sample tested
 |
|
The overall ZIP δ synergy score was based on the entire range of doses tested, and the most synergistic area score was based on the specific concentrations that had the highest synergy for each AML and DLBCL sample tested. Red shading indicates synergistic ZIP scores >0, green shading indicates antagonistic ZIP scores <0. AML samples that responded to venetoclax in the clinic are indicated by an orange font. AML samples that were primary induction refractory to venetoclax in the clinic are indicated by a lavender font.
To further substantiate this treatment combination in AML, we tested the effects of treatment with eltanexor-venetoclax in a venetoclax-resistant AML PDX model (supplemental Table 1; patient AML-008). Seven weeks after injection, human CD45+ chimerism was established in the peripheral blood (Figure 5D), and the mice were given vehicle control, eltanexor (7.5 mg/kg), venetoclax (25 mg/kg), or eltanexor-venetoclax combined starting at week 8 for 5 days per week for 6 weeks. At 93 days posttransplantation, the experiment was terminated to determine the levels of leukemia engraftment in the bone marrow and spleen. As with the MV-4-11 model, gross examination revealed a decrease in spleen weight in all treatment groups without affecting total body weight (supplemental Figure 10A-C). Although treatment with this dose of venetoclax displayed minimal reductions in human CD45+ cells in the peripheral blood and spleen, it was less effective in the bone marrow. Treatment with single-agent eltanexor led to slightly more enhanced effects; however, treatment with eltanexor-venetoclax combined revealed dramatic reductions in human CD45+ cells (Figure 5D-E; supplemental Figure 10B-C). Immunohistochemistry analysis of human CD45 (hCD45) cells in the bone marrow and splenic tissue corroborated the findings assessed by flow cytometry for all of the treatment groups (Figure 5F). Pathologic review of sections stained with hematoxylin and eosin indicated no evidence of histologic abnormalities that would be of concern for off-target drug-related effects in the liver, lung, heart, and kidneys of mice treated for 6 weeks with eltanexor (7.5 mg/kg), venetoclax (25 mg/kg), or eltanexor-venetoclax combined (supplemental Figure 10E).
Discussion
Most critical tumor suppressors and oncogenic mRNAs are only exported from the nucleus by XPO1; thus, leveraging this mechanism is of great interest as a therapeutic option. The targeting of XPO1 with the SINE compounds, selinexor, or eltanexor has led to responses in refractory and aggressive hematologic malignancies in phase 1/2 studies.11,12,22,56 BCL2 inhibition with the BH3 mimetic venetoclax targets a hallmark of cancer—evasion of apoptosis—with sharp focus. The recent success of venetoclax in the clinic, however, has been most cogent with combination therapy that leverages standard-of-care therapy (eg, decitabine, 5′azacitidine, cytarabine, rituximab) as priming agents for venetoclax.35,36,38 The combination of SINE and venetoclax has not been fully explored; recently, however, investigators have illustrated that selinexor enhances cell death in AML cell lines and patient samples treated with venetoclax and also in glioblastoma PDX models and cell lines treated with ABT-263 (inhibitor of BCL2, BCL-xL, and BCL-W).57,58 Here, we have shown that the combination of SINE compounds and venetoclax effectively and synergistically induces apoptosis in AML, which leads to a decrease in cell growth and diminished in vivo leukemia burden, with almost complete clearance in the bone marrow, spleen, and peripheral blood of animals that have received a transplantation. The combination of SINE compounds and venetoclax also shrank tumors and extended survival in all tested lymphoma models and even resulted in lymphoma regression and stabilization within an advanced large tumor model. We have also shown that low doses of the SINE-venetoclax combination effectively reduced AML blasts while sparing most of the normal blasts. Thus, in the clinic, a treatment regimen with a SINE-venetoclax combination could provide the benefits of all oral therapies while minimizing effects on normal hematopoiesis seen when venetoclax is combined with DNA methyltransferase inhibitors or low-dose cytarabine (cytotoxic partners approved for use) in the clinic.
SINE compounds have pleiotropic effects on cells because of myriad cargo proteins that are affected, but one possible mechanism to explain the synergy of the SINE-venetoclax combination is related to the ability of SINE molecules to alter critical proteins involved in the regulation of apoptosis, particularly p53 and MCL1. Increased p53 protein in response to treatment with SINE compounds in both AML and DLBCL cells is important in the context of this combination, because p53 activation has been shown to be synthetically lethal when combined with BCL2 inhibition in both models of p53 overexpression and in PDXs with pharmacologic inhibitors of p53 degradation.49 Activation of p53 also leads to proteasomal degradation of MCL1 through activation of GSK3,49 perhaps muting any MCL1 upregulation which may occur as a consequence of exposure to venetoclax and may further confer the synergy seen in our experiments. The lack of quantitative change in p53, despite marked synergy with the SINE-venetoclax combination, along with negligible alterations to response with TP53 knockdown suggests a potential p53-independent synergy of SINE and venetoclax, and this might be clinically relevant, given the approval of venetoclax and considerable response of 17p del CLL (eg, TP53 deleted).36,59 Blockade of MCL1 not only kills cells that have upregulated MCL1, but it may also re-sensitize those venetoclax-resistant cells to venetoclax.32,41,60,61 In a p53-independent manner, the SINE-venetoclax combination seems to limit the production and activity of MCL1. Inhibition of XPO1 prevents eIF4E from enhancing the translation of BCL2 and MCL1 mRNA.6,45-47 Thus, there are several mechanisms for treatment with SINE compounds to decrease MCL1 protein, which may ultimately be shown to alleviate tumor resistance that arises via upregulation of MCL1 in the setting of treatment with venetoclax.
However, just as we saw minimal effects with TP53 knockdown, overexpression of MCL1 did not completely abrogate the synergistic effects of treatment with the SINE-venetoclax combination.58 Thus, we believe the superior anti-tumor effects of the SINE-venetoclax combination is a result of the collective activity of several proteins affected by SINE,1-4,6 which have alternate roles depending on the cellular context. The combination of SINE compounds and venetoclax may illustrate an opportunity to overcome cancer heterogeneity, and the precise mechanism of action for a given context may be dependent on any of the large number of carrier proteins affected by SINE compounds.1-4,6 To this end, further investigation is needed.
We and others have recently noted specific activity with novel, selective MCL1 inhibitors in AML, synergy between these inhibitors and venetoclax, and the rescue of venetoclax resistance with these inhibitors.32,33,44 The combination of venetoclax with selective MCL1 inhibitors in the future is a likely potential strategy for treating AML, as trials with selective MCL1 inhibitors continue. However, potent inhibition of both MCL1 and BCL2 in normal cells remains a concern,32,62 and alternative strategies with surrogate anticancer agents which may reduce MCL1 may have additional value.61 Perhaps our most promising finding was that AML patient samples that were primary refractory to venetoclax in the clinic still responded synergistically to treatment with the SINE-venetoclax combination ex vivo, including a PDX that used a venetoclax-resistant patient sample. This warrants further investigation into the effect of SINE compounds in the context of circumventing de novo venetoclax resistance. The data presented here provide a proof of principle for the design of a clinical trial combining SINE molecules and venetoclax in treating high-risk hematologic malignancies; a clinical trial testing selinexor and venetoclax in combination in AML and DLBCL is underway (NCT03955783).
Questions about protocols or reagents may be addressed to Michael R. Savona (michael.savona@vanderbilt.edu).
Acknowledgments
The Vanderbilt-Ingram Cancer Center Hematologic Malignancy Tumor Bank was critical in completion of this work.
This work was supported by the Biff Ruttenberg Foundation, the Adventure Allie Grant, the E. P. Evans Foundation, as well as Karyopharm Therapeutics. The Vanderbilt-Ingram Cancer Center is supported by a National Institutes of Health (NIH), National Cancer Institute (NCI) grant P30 CA068485-19. The REDCap database tool is supported by grant UL1 TR000445 from the NIH, National Center for Advancing Translational Sciences. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource, which is supported by the Vanderbilt-Ingram Cancer Center, NIH, NCI (P30 CA68485) and the Vanderbilt Digestive Disease Research Center, NIH, National Institute of Diabetes and Digestive and Kidney Diseases (DK058404). M.R.S. is a Leukemia and Lymphoma Society Clinical Scholar.
Authorship
Contribution: M.A.F. designed and performed experiments, analyzed data, performed statistical analysis, and wrote the manuscript; S.Y.F., H.C., and W.S. designed and performed experiments, analyzed data, and edited the manuscript; M.P.A., A.E.G., L.D.F., H.E.R., T.K., C.A., S.D., M.B., M.T.V., and A.C.S. performed the experiments and edited the manuscript; Y.L., E.B., and S.S. designed experiments and edited the manuscript; S.S. designed selinexor; E.B. designed eltanexor; M.R.S. designed experiments, analyzed data, wrote the manuscript, and supervised the study; and all authors reviewed drafts of the manuscript and approved the final version of the manuscript.
Conflict-of-interest disclosure: S.Y.F., H.C., C.A., and S.D. are employed by and have equity ownership in Karyopharm Therapeutics. T.K., W.S., Y.L., and E.B. are employed by and have equity ownership in Karyopharm Therapeutics and have relevant patents. S.S. is employed by and has equity ownership in Karyopharm Therapeutics, has relevant patents, and is on the Karyopharm Therapeutics scientific advisory board. M.B. receives research funding from AbbVie and Karyopharm Therapeutics. M.R.S. consults for and serves on the scientific advisory board and had equity in Karyopharm Therapeutics; receives research funding from Astex, Incyte, Millennium Pharmaceuticals, and TG Therapeutics; serves as a consultant and is on the advisory board and monitoring committees for AbbVie, Astex, Celgene, Gilead, Incyte, Karyopharm Therapeutics, Millennium Pharmaceuticals, Ryvu, and TG Therapeutics; and has patents and royalties from Boehringer-Ingelheim. The remaining authors declare no competing financial interests.
Correspondence: Michael R. Savona, Vanderbilt University School of Medicine, 777 Preston Research Building, 2220 Pierce Ave, Nashville, TN 37232; e-mail: michael.savona@vanderbilt.edu.

