

Key Points
Pneumolysin induces pores in platelets, which renders platelets nonfunctional.
Polyvalent immunoglobulins prevent platelets from damage by pneumolysin.

Abstract
Community-acquired pneumonia by primary or superinfections with Streptococcus pneumoniae can lead to acute respiratory distress requiring mechanical ventilation. The pore-forming toxin pneumolysin alters the alveolar-capillary barrier and causes extravasation of protein-rich fluid into the interstitial pulmonary tissue, which impairs gas exchange. Platelets usually prevent endothelial leakage in inflamed pulmonary tissue by sealing inflammation-induced endothelial gaps. We not only confirm that S pneumoniae induces CD62P expression in platelets, but we also show that, in the presence of pneumolysin, CD62P expression is not associated with platelet activation. Pneumolysin induces pores in the platelet membrane, which allow anti-CD62P antibodies to stain the intracellular CD62P without platelet activation. Pneumolysin treatment also results in calcium efflux, increase in light transmission by platelet lysis (not aggregation), loss of platelet thrombus formation in the flow chamber, and loss of pore-sealing capacity of platelets in the Boyden chamber. Specific anti-pneumolysin monoclonal and polyclonal antibodies inhibit these effects of pneumolysin on platelets as do polyvalent human immunoglobulins. In a post hoc analysis of the prospective randomized phase 2 CIGMA trial, we show that administration of a polyvalent immunoglobulin preparation was associated with a nominally higher platelet count and nominally improved survival in patients with severe S pneumoniae–related community-acquired pneumonia. Although, due to the low number of patients, no definitive conclusion can be made, our findings provide a rationale for investigation of pharmacologic immunoglobulin preparations to target pneumolysin by polyvalent immunoglobulin preparations in severe community-acquired pneumococcal pneumonia, to counteract the risk of these patients becoming ventilation dependent. This trial was registered at www.clinicaltrials.gov as #NCT01420744.
Introduction
Community-acquired pneumonia by primary or secondary infection with Streptococcus pneumoniae (S pneumoniae; the pneumococcus) is one of the most frequent severe infections associated with high mortality.1,2 Patients are at risk of developing acute respiratory distress syndrome requiring mechanical ventilation. A hallmark of acute respiratory distress syndrome is extravasation of protein-rich fluid into the pulmonary tissue, for example, when the pore-forming toxin pneumolysin alters the alveolo-capillary barrier.3 In vivo concentrations of pneumolysin occurring during acute pneumonia or invasive infections have not been adequately determined. It can be assumed that the local concentrations differ greatly from circulating pneumolysin concentrations due to dilution in the flowing blood. In an experimental pneumococcal pneumonia mouse model, pneumolysin concentrations correlated with the number of bacteria and the highest concentration measured in the peripheral blood was ∼1 ng/mL. This sublytic pneumolysin concentration induced tissue damage including cardiomyocyte injury and dysfunction, and was suggested to be involved in apoptosis of cells of the host immune system.4,5 The only organ in which pneumolysin is not diluted by the blood flow is the cerebrospinal fluid in patients with pneumococcal meningitis. In this situation, pneumolysin concentrations of 0.85 ng/mL to 180 ng/mL or 10 µg/mL to 30 µg/mL have been measured in the cerebrospinal fluid.6,7
Platelets play a major role in maintaining the endothelial barrier.8 We show that pneumolysin secreted by pneumococci renders platelets nonfunctional. These platelets can no longer seal gaps. Polyvalent immunoglobulins prevent this loss of platelet function. In addition, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)9,10 and influenza virus are also causing severe virus-induced pneumonia. A subset of these patients might suffer from pneumococcal coinfections, which in part may add on to their ventilation needs.11
Methods
Ethics
The CIGMA study was conducted in accordance with the International Council for Harmonization, Good Clinical Practice standards, and the Declaration of Helsinki, and with the approval of local institutional review boards/independent ethics committees. All patients (or their representatives) provided written informed consent.
The use of whole blood and washed platelets from healthy adult individuals was approved by the Ethics Committee of the University Medicine Greifswald (BB 044/18). All volunteers gave written informed consent in accordance with the Declaration of Helsinki. All experiments were carried out in accordance with the approved guidelines.
Antibodies and reagents
We used the following antibodies and reagents: mouse monoclonal anti-pneumolysin (Abcam, Cambridge, MA), rabbit polyclonal anti-pneumolysin antibody (Davids Biotechnologie GmbH, Regensburg, Germany), mouse polyclonal anti-enolase antibody (routine immunization of mice with heterologously expressed enolase), IRDye 800CW goat anti-mouse immunoglobulin G (IgG) antibody (Abcam), IRDye 680RD goat anti-rabbit IgG antibody (Abcam), phycoerythrin (PE)-Cy5–labeled monoclonal mouse anti-human CD62P, fluorescein isothiocyanate (FITC)-labeled mouse PAC-1 antibodies recognizing activated αIIbβIII (CD41/CD61), the RealTime-Glo MT Cell Viability Assay (Promega, Madison, WI), human polyvalent immunoglobulin preparations (pharmaceutical human IgG; IgG-enriched Privigen; CSL Behring, Marburg, Germany) and trimodulin [Biotest, Dreieich, Germany]), FITC-labeled mouse anti-human CD42a (BD Biosciences, Franklin Lakes, NJ), monoclonal mouse anti-human α-tubulin antibody (clone DM1A; Sigma-Aldrich, St. Louis, MO), ATTO 488–labeled Phalloidin (ATTO-TEC GmbH, Siegen, Germany), Arg-Gly-Asp-Ser (RGDS; Sigma-Aldrich), Alexa Fluor 647–labeled monoclonal mouse anti-human CD62P (P-selectin) antibody (clone AK4; BioLegend, San Diego, CA), Alexa Fluor 647-labeled goat anti-mouse IgG (GAMIG AF-647) (Abcam), and Triton X-100 (Sigma-Aldrich).
Platelet preparation
We purified platelets from acid citrate dextrose solution A (ACD-A) anticoagulated whole blood from healthy donors who did not take antiplatelet drugs or nonsteroidal anti-inflammatory drugs (NSAIDs) and used the platelets of the same volunteers for repeated experiments. We prepared platelets as described.12 In brief, we washed platelet-rich plasma (PRP) twice with Tyrode buffer containing 0.35% bovine serum albumin (BSA), 0.1% glucose, 2.5 U/mL apyrase, 1 U/mL hirudin, pH 6.3; resuspended the final platelet pellet in a bicarbonate-based suspension buffer containing 0.35% BSA, 0.1% glucose, 0.212 M MgCl2, 0.196 M CaCl2, pH 7.2; and adjusted them to 300 000 platelets per microliter.13
Flow cytometry–based platelet-activation assay
We performed platelet-activation assays as described.13 Briefly, we incubated washed human platelets in Tyrode buffer containing Ca2+ and Mg2+ with phosphate-buffered saline (PBS), 20 µM thrombin receptor activator peptide 6 (TRAP-6), pneumolysin, or pneumococci (D39, TIGR4, or their pneumolysin-free mutants D39Δply and TIGR4Δply).14,15 We did grow bacteria to the mid-log exponential phase before incubating 1.8 × 106 bacteria with 9 × 106 platelets (ratio bacteria platelets 1:5) for 2 or 3 hours. We incubated platelets with pneumolysin at different concentrations or the pneumolysin mutants for 10 minutes. We measured CD62P expression using a mouse monoclonal PE-Cy5–conjugated CD62P antibody. In addition, we determined αIIbβIII (CD41/CD61) activation using the FITC-labeled mouse PAC-1 antibody. After 10 minutes of incubation of platelets with the antibodies at room temperature (RT), we fixed platelets with paraformaldehyde (PFA)/PBS (pH 7.4) at a final concentration of 2% for 20 minutes at RT and measured them after 2 washing steps (700g, 7 minutes) using a FACSCalibur (Becton Dickinson) flow cytometer and CellQuestPro 6.0 or the Cytomics FC 500 (Beckman Coulter) and CXP 2.2 software. We then predefined by forward-sideward scatter a platelet gate based on measurements with CD61+ platelets and analyzed in the gated region 20 000 events for fluorescence. We then calculated the value for platelet activation as the geometric mean fluorescence intensity (GMFI) of the gated population multiplied by the percentage of CD62P+-labeled platelets.
Field emission scanning electron microscopy
We performed field emission scanning electron microscopy (FESEM) identical to the protocol described by Binsker et al.13 Briefly, we incubated washed platelets with pneumococci (60 minutes) or pneumolysin proteins (10 minutes) followed by fixation with 1% formaldehyde at RT. Samples were then centrifuged at 2000g for 2 minutes, washed with TE buffer (20 mM Tris-HCl, 2 mM EDTA, pH 6.9), and the resulting pellet was resuspended in 50 µL of TE buffer. Fifty microliters of resuspended samples were placed onto poly-l-lysine–coated cover slips (12 mm in diameter), fixed with 1% glutaraldehyde in TE buffer for 10 minutes, washed with TE buffer, critical point dried with acetone (CPD 300; Leica), and sputter coated with gold-palladium (SCD 500; Bal-Tec). For imaging in a field emission scanning electron microscope (Zeiss Merlin, Oberkochen, Germany), we used the Everhart-Thornley SE detector alone or together with the Inlens SE detector at a 75:25 ratio at an acceleration voltage of 5 kV and SmartSEM software 6.06 or 5.05.
Pneumolysin production, platelet treatment, and neutralization
For all platelet experiments, we used recombinant cytolytic active pneumolysin and mutants of pneumolysin, pneumolysinC428G without cytolytic activity, and pneumolysinW433F with ∼10% cytolytic activity in PBS. We amplified the pneumolysin gene by polymerase chain reaction (PCR) using genomic DNA from Streptococcus pneumoniae TIGR4 using the forward primer 370 N-Ply 5′-CGGGATCCGCAAATAAAGCAGTAAATGAC-3′ and reverse primer 371 C-Ply 5′-GCGGTACCTAGTCATTTTCTACCTGAG-3′. We ligated the BamHI-digested PCR product into the BamHI- and EcoRV-digested vector pASK-IBA5 (IBA). The resulting recombinant plasmid pKK2 was used for site-directed mutagenesis with the QuikChange XL site-directed mutagenesis kit (Agilent Technologies). For the amino acid exchange, the following primer combinations were used for an inverse PCR of pKK2: primer 453 PlyW433F+ CCGGGCTAGCCTTCGAATGGTGGCGTACGG-3′ and 454 PlyW433F- 5′-CACCATTCGAAGGCTAGCCCGGTACACTCTC-3±′, 455 PlyC428G+ 5′-GAGAGGGTACCGGGCTAGCCTGGGAATGGTGGC-3′, and 456 PlyC428G- 5′-CCCAGGCTAGCCCGGTACCCTCTCTAATTTTGA-3′. Escherichia coli DH5α was transformed with the resulting PCR products, after digestion with DpnI to get rid of the template.
For protein production, we cultured E coli SCS1 containing the expression plasmids for pneumolysin in SB (super broth) medium at 30°C with shaking.13 At OD600 2.0, protein expression was induced for 3 hours at RT with 1 mM anhydrotetracycline. After cell lysis for protein purification, we performed affinity chromatography using a StrepTrap HP column according to the manufacturer’s instructions (GE Healthcare). We dialyzed pneumolysin proteins against PBS (pH 7.4) overnight at 4°C and determined protein concentrations using a Bradford assay. We determined cytolytic activity of purified pneumolysin proteins by the hemolysis activity test as described.14 In brief, we incubated ACD-A blood from healthy human volunteers with pneumolysinWT, pneumolysinC428G, and pneumolysinW433F for 10 minutes at 37°C in a 96-well plate (U-bottom). After incubation, we centrifuged the plate and monitored formation of the erythrocyte sediment.
In platelet-activation assays with pneumolysin, we treated platelets for 4 minutes with 300 µg/mL, 3.0 µg/mL, 300 ng/mL, 30 ng/mL, or 3.0 ng/mL pneumolysin followed by 5-minute treatment with 20 µM TRAP-6. In neutralization experiments, we preincubated pneumolysin for 20 minutes at RT with 1 mg/mL human IV immunoglobulin (pharmaceutical human IgG) (IgG-enriched Privigen; CSL Behring, Marburg, Germany), 7.5 µg/mL mouse monoclonal anti-pneumolysin (Abcam), or 10 µg/mL rabbit polyclonal anti-pneumolysin antibodies.
Immunofluorescence staining of platelets
Three million platelets (300 000 cells per microliter) were incubated in Tyrode buffer (resting platelets) or treated with TRAP-6 (20 µM, control for activation), Triton X-100 (0.1%, control for detergent-induced pore formation) or pneumolysin (3, 5, 20, 30, 50, 100, 300 ng/mL) for 10 minutes at 37°C. Samples were fixed in 2% PFA for 20 minutes and subsequently spun on microscopy slides using the Cytospin system (Thermo Fisher). Slides were washed 3 times in PBS pH 7.2. Platelets were then incubated with anti-CD62P-AF647 (1:100) and phalloidin-ATTO 488 (20 pM) or monoclonal mouse anti-α-tubulin antibody (clone DM1A, 1:100) for 2 hours at RT in the dark. Afterward, slides were washed 3 times in PBS pH 7.2. Anti-CD62P-AF647 and phalloidin ATTO 488–stained platelets were then covered by 20 µL of fluorescent mounting medium (ROTI Mount FluorCare HP19; Carl Roth GmbH, Karlsruhe, Germany) and a coverslip. Platelets incubated with monoclonal mouse anti-α-tubulin antibody (used as control for a strictly intracellular protein) were stained with GAMIG-AF647 (1:750) secondary antibody and phalloidin-ATTO 488 (20 pM) for 2 hours at RT in the dark, again washed 3 times in PBS pH 7.2, and subsequently covered by 20 µL of fluorescent mounting medium and a glass coverslip. Confocal laser microscopy was performed on a Leica SP5 confocal laser scanning microscope (Leica, Wetzlar, Germany) equipped with HCX PL APO λ blue 40.0×/1.25 oil UV objective. For image acquisition, ATTO 488 and AF647 were excited by argon (488 nm) and helium-neon (HeNe; 633 nm) laser lines selected with an acousto-optic tunable filter (AOTF) and fluorescence emission was collected between 505 and 515 nm and 640 and 655 nm, respectively, on hybrid detectors (HyDs).
Assessment of CD62P immunofluorescence signal intensities and localization and α-tubulin staining was performed by measuring the line profile (5 µm length and 1 µm width) of nonsaturated grayscale fluorescence intensities (pixel values) of immunofluorescent probes across individual platelets (≥20) in confocal images. To provide further evidence for intracellular staining of CD62P, we performed confocal Z-stacks of platelets and created orthogonal views and 3-dimensional (3D) rendering.
Cell culture and Boyden chamber assays
We transferred 150 µL of washed platelets (300 000/µL) in Tyrode buffer containing 0.212 M MgCl2, 0.196 M CaCl2 into the upper well of a Boyden chamber (6.5-mm Transwell with 3.0-µm Pore Polycarbonate Membrane Insert; Corning). Then we added pneumolysin to the upper and lower chamber in the same concentration each (300 ng/mL, 30 ng/mL, and 3 ng/mL final) and incubated for 45 minutes at 37°C. Ca2+, which can be taken up by cells and has been shown to be necessary for repair of pneumolysin-induced pores in eukaryotic membranes,16 was present during the experiment. In a subset of experiments, a pneumolysin-inhibiting monoclonal mouse antibody (7.5 µg/mL), polyclonal rabbit anti-pneumolysin antibodies (10 µg/mL), and a pharmaceutical human IgG (IgG-enriched Privigen) preparation (1 mg/mL) were added. We then transferred the inserts into new wells containing 0.9% NaCl, and BSA-FITC (ThermoFisher) was pipetted into the upper chamber at a final concentration of 0.25 mg/mL and incubated for 10 minutes at RT in darkness. We determined platelet pore-sealing capacity by measuring the fluorescence signal of BSA-FITC in the flow-through using a Fluoroskan Ascent FL fluorimeter.
Light transmission aggregometry
We resuspended washed platelet-suspension buffer and added fibrinogen to a final concentration of 2.25 mg/mL. In some experiments, the following were added: RGDS peptides to a final concentration of 1.16 mM, or a pneumolysin-inhibiting monoclonal mouse antibody (7.5 µg/mL), polyclonal rabbit anti-pneumolysin antibodies (10 µg/mL), or a pharmaceutical human IgG preparation (1 mg/mL; Privigen, CSL Behring). After transfer to the aggregometer cuvette, different concentrations of pneumolysin were added to the platelet suspension after 15 seconds. We measured platelet aggregation as a decrease in turbidity of the medium with an APACT4 aggregometer at 500 rpm, 37°C (Haemochrom) applying APACT LPC software. In some experiments, we added 20 µM TRAP-6 after 240 seconds and continued measurement for a further 200 seconds.
Live/dead staining
For measurement of cell viability, we used the RealTime-Glo Cell Viability Assay (Promega) and measured viability for 30 minutes. We used the ability of a cell to reduce a substrate as a mean for viability. We mixed the assay substrate 1:1 with the twofold concentration of pneumolysin. In a subset of experiments, a pneumolysin-inhibiting monoclonal mouse antibody (7.5 µg/mL), polyclonal rabbit anti-pneumolysin antibodies (10 µg/mL), or a pharmaceutical human IgG preparation (1 mg/mL; IgG-enriched Privigen; CSL-Behring) were added. Then, we added the pneumolysin-substrate mixture to washed human platelets in a 96-well plate in duplicates. After 1 minute of incubation, we started shaking the plate with 300 rpm for 3 seconds and measured relative luminescence units (RLUs) using a microtiter plate reader. We repeated shaking and measurement of luminescence every 60 seconds until a total measurement time of 30 minutes. Sample values of luminescence represent the mean of the duplicates subtracted by blank (Tyrode buffer without platelets) of 6 independent experiments.
Release of intracellular calcium
We detected the release of Ca2+ from internal stores to the cytoplasm by fluorescent labeling of free intracellular Ca2+ using Fluo-4-AM (ThermoFisher). We resuspended platelets in PBS without MgCl2 and CaCl2 (pH 7.4), adjusted them to 150 000 platelets per microliter, and stained them with Fluo-4-AM for 30 minutes in the dark at RT. Buffers needed to be calcium free as diffusion of extracellular calcium or fluorescent dye through the pores would have caused artifacts. After a 1/2 dilution in PBS, we carried out baseline measurements for 15 seconds. Afterward, we stimulated platelets with a final concentration of 300 ng/mL, 30 ng/mL, and 3.0 ng/mL pneumolysin. In a subset of experiments, a pneumolysin-inhibiting monoclonal mouse antibody (7.5 µg/mL), polyclonal rabbit anti-pneumolysin antibodies (10 µg/mL), or a pharmaceutical human IgG preparation (1 mg/mL; IgG-enriched Privigen; CSL-Behring) were added. We measured free Ca2+ with a Fluoroskan Ascent FL fluorometer (ThermoFisher) over 7 minutes. In some experiments, we added TRAP-6 at a final concentration of 20 µM after 250 seconds and the measurement was carried out for another 200 seconds.
Thrombus formation under shear flow
We incubated hirudinized whole blood (1 mL) with 3 ng/mL, 30 ng/mL, and 300 ng/mL pneumolysin and pneumolysinC428G (without cytolytic activity) and pneumolysinW433F (with ≈10% cytolytic activity) at 300ng/mL final concentration in whole blood for 10 minutes. In a subset of experiments, pharmaceutical human IgG preparation (1 mg/mL; IgG-enriched Privigen; CSL-Behring) was added to hirudinized whole blood either in the absence (human IgG control) or in the presence of 300 ng/mL pneumolysin. We performed thrombus formation assays at a wall shear rate of 1000 s−1 on collagen-passivated surfaces (200 µg/mL HORM collagen type I from horse tendon; Nycomed) in a microfluidic parallel platelet flow chamber (on µ-Slide VI 0.1 with physical dimensions: 1 mm width, 100 µm height, and 17 mm length [Ibidi]). To visualize thrombus formation, prior to perfusion, platelets were labeled with monoclonal antibody (mAb) CD42a-FITC (0.125 µg/mL). We performed time-lapse confocal imaging at intervals of 10 seconds per image on a Leica SP5 confocal laser scanning microscope (Leica) equipped with a water immersion HC PL APO 20×/0.75 IMM CS2 objective. FITC was exited at 488 nm with an argon laser line selected with AOTF; fluorescence emission was collected between 505 and 515 nm on a HyD. We performed quantitative assessment of platelet adhesion and thrombus formation to obtain the percentage area covered by thrombi over time by computational image analysis using the surfaces creation wizard algorithm in Bitplane Imaris version 7.65. (Oxford Instruments, Abingdon, United Kingdom). Experiments were performed according to International Society on Thrombosis and Haemostasis Scientific and Standardization Committee (ISTH SSC) subcommittee on Biorheology recommendations.14
Hemolysis assay
To test the cytolytic activity of pneumolysin proteins, the hemolysis assay was performed as described recently.15 In a subset of experiments, a pneumolysin-inhibiting monoclonal mouse antibody (7.5 µg/mL), polyclonal rabbit anti-pneumolysin antibodies (10 µg/mL), or a pharmaceutical human IgG preparation (1 mg/mL; IgG-enriched Privigen; CSL-Behring) were added.
To analyze the capacity of trimodulin to neutralize pneumolysin, we incubated different concentrations of trimodulin with 300 ng/mL pneumolysin for 30 minutes at 37°C. Afterward, we added 1.5% human whole blood in PBS for 60 minutes at 37°C. Following centrifugation, we measured the supernatant for hemoglobin content by spectrophotometry at 450 nm. As positive control, we used 1% Triton X-100; as negative control, we used Dulbecco PBS.
Quantification of pneumolysin in pneumococci and culture supernatants
S pneumoniae D39, D39Δply, TIGR4, and TIGR4Δply were grown until mid-log phase, harvested, and resuspended in PBS containing 30% Tyrode buffer (without BSA). Generation of the pneumolysin mutants has been described recently.15 After 2 hours and 3 hours of incubation in PBS/Tyrode buffer at 37°C, pneumococci and supernatants were collected for immunoblotting. A total of 1 × 108 bacteria and the respective trichloroacetic acid precipitated supernatant of 1 × 108 bacteria were run on a 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). In addition, a serial dilution of recombinant pneumolysin protein was used as standard. The samples were blotted on a nitrocellulose membrane and, after blocking, polyclonal primary antibodies raised in rabbit or mouse and fluorescent-labeled secondary antibodies were used to detect pneumolysin or enolase using the Odyssey CLx scanner (Li-Cor). Pneumolysin signals in the supernatants were normalized using the enolase signal of the respective bacterial lysate. Normalization was performed using Image Studio software. Calculation of the pneumolysin amount based on the pneumolysin standard curve was performed with Microsoft Excel (Office package 2016). After normalization and quantification, the immunoblot images were adjusted for brightness and contrast using Photoshop CS5.
Patient analysis
We investigated the effect of immunoglobulins on platelet numbers in patients with S pneumoniae infection with data generated in the CIGMA study (NCT01420744).17 The patients were treated with trimodulin (182.6 mg/kg) or placebo for 5 consecutive days. Trimodulin is a human polyvalent immunoglobulin preparation containing 45 to 55 mg/mL human plasma immunoglobulin proteins; it is composed of ∼23% IgM, ∼21% IgA, and ∼56% total IgG. In 100 of the 160 patients enrolled into the study, the causative pathogens (bacterial and/or viral) were identified.
Statistics
We performed statistical analysis using GraphPad Prism (version 5.01), unless otherwise indicated. We show the data as scatter plots and include median, minimal, and maximal values including median and interquartile range. We analyzed the data using the nonparametric Friedman test followed by a Dunn multiple comparison posttest. We considered P < .05 to be statistically significant.
Results
Pneumococci induce platelet staining for CD62P
We incubated platelets with wild-type and pneumolysin-deficient pneumococci. Wild-type pneumococci induced staining for the platelet activation marker CD62P (P-selectin; supplemental Figure 1). This was strongest for the strain TIGR4, less pronounced for strain D39, which produces less pneumolysin, and lowest for pneumolysin-deficient ply mutants. We therefore expected that pneumococci preactivate platelets and render them more reactive. However, in the presence of wild-type TIGR4 pneumococci, platelets were no longer reactive to costimulation with TRAP-6 (a potent platelet thrombin receptor agonist), whereas D39 and pneumolysin-deficient pneumococci still allowed additional platelet activation by TRAP-6. This suggested that pneumolysin interferes with platelet reactivity.
Pneumolysin induces pores in the platelet membrane, Ca2+ efflux, and platelet lysis
When we incubated platelets with purified pneumolysin, we observed a similar pattern as described in the previous paragraph for incubation of platelets with wild-type pneumococci (Figure 1A-C refers to experiments with neutralizing antibodies explained in detail below in “Antibodies and polyvalent immunoglobulins inhibit the effects of pneumolysin on platelets in vitro”). In contrast, pneumolysin proteins without or with low cytolytic activity failed to induce CD62P expression (Figure 1D-E) and integrin activation (supplemental Figure 2A-D) and platelets remained responsive to TRAP-6. To better understand the effects of pneumolysin, we visualized platelets incubated with wild-type or pneumolysin-deficient pneumococci by scanning electron microscopy.
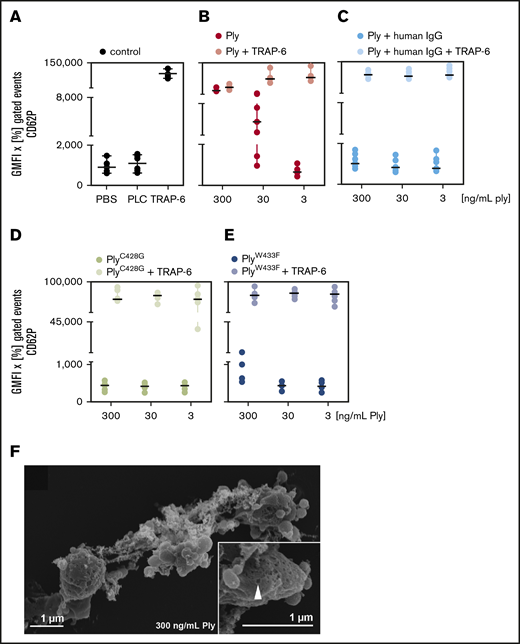
CD62P expression of platelets by pneumolysin is caused by pore formation. Washed platelets of a defined set of 6 donors were incubated with various concentrations of pneumolysin (Ply). CD62P was detected by flow cytometry using antibodies against CD62P (P-selectin). The data are presented as GMFI of the positive gated events multiplied with the percentage of positive gated events in the dot plots. (A) PBS (gray) and phospholipase C (PLC; gray) from Staphylococcus aureus known to not activate platelets13 were used as negative controls and 20 µM TRAP-6 (gray) as positive control. (B) Pneumolysin (red; ng/mL) caused CD62P expression and dose-dependently inhibited an additional response to TRAP-6 (black). (C) Polyvalent human immunoglobulins (human IgG; Privigen) neutralized the effect of pneumolysin (pneumolysin plus human IgG = light blue) (to enable comparison with the experiments without immunoglobulins, the data are shown here, although they are presented in the text at the end of “Results”). (D) PneumolysinC428G without lytic activity (brown) did not activate platelets or impaired the response to TRAP-6 and (E) pneumolysinW433F with ∼10% lytic activity (purple) had a very minor effect only at 300 ng/mL. (F) Visualization of pore formation in the platelet membrane by pneumolysin by scanning electron microscopy. Platelets are altered in their shape and formed vesicles but not pseudopodias. At the left side, a platelet with pores can be seen. Inset, a higher magnification of the platelet indicating a pore by an arrow.
CD62P expression of platelets by pneumolysin is caused by pore formation. Washed platelets of a defined set of 6 donors were incubated with various concentrations of pneumolysin (Ply). CD62P was detected by flow cytometry using antibodies against CD62P (P-selectin). The data are presented as GMFI of the positive gated events multiplied with the percentage of positive gated events in the dot plots. (A) PBS (gray) and phospholipase C (PLC; gray) from Staphylococcus aureus known to not activate platelets13 were used as negative controls and 20 µM TRAP-6 (gray) as positive control. (B) Pneumolysin (red; ng/mL) caused CD62P expression and dose-dependently inhibited an additional response to TRAP-6 (black). (C) Polyvalent human immunoglobulins (human IgG; Privigen) neutralized the effect of pneumolysin (pneumolysin plus human IgG = light blue) (to enable comparison with the experiments without immunoglobulins, the data are shown here, although they are presented in the text at the end of “Results”). (D) PneumolysinC428G without lytic activity (brown) did not activate platelets or impaired the response to TRAP-6 and (E) pneumolysinW433F with ∼10% lytic activity (purple) had a very minor effect only at 300 ng/mL. (F) Visualization of pore formation in the platelet membrane by pneumolysin by scanning electron microscopy. Platelets are altered in their shape and formed vesicles but not pseudopodias. At the left side, a platelet with pores can be seen. Inset, a higher magnification of the platelet indicating a pore by an arrow.
We observed binding of both wild-type (supplemental Figure 3C-D) and pneumolysin-deficient pneumococci (supplemental Figure 3A-B) to platelets, but only wild-type pneumococci induced pores (supplemental Figure 3C-D). Controls are shown in supplemental Figure 3E-F. We observed pore formation with diameters of 40 to 50 nm in platelets when we added purified pneumolysin at concentrations of 300 µg/mL (supplemental Figure 4A), 300 ng/mL (Figure 1F; supplemental Figure 4B), 30 ng/mL (supplemental Figure 4C) and 3.0 ng/mL (supplemental Figure 4D), but not when we applied inactive pneumolysins (supplemental Figure 4E-F). The pneumolysin concentrations causing pores correspond to the concentrations causing hemolysis in erythrocytes (supplemental Figure 5A-B).
We then assessed the concentration of pneumolysin in bacterial culture supernatants by SDS-PAGE (supplemental Figure 5C). The intensities of the pneumolysin protein bands correspond to 2.677 ± 0.871 ng/mL pneumolysin for strain TIGR4 and 1.834 ± 0.261 ng/mL pneumolysin for strain D39 (supplemental Figure 5D) after 3 hours of incubation.
We next addressed the consequences of pore formation on platelet function. When we incubated washed platelets with 300 ng/mL pneumolysin, we observed an immediate release (2.5-fold) of Ca2+ (Figure 2A; supplemental Figure 6A), consistent with the findings of others,18 and an increase in platelet aggregation (up to 50%) as measured by a change in light transmission of the platelet suspension (Figure 2B; supplemental Figure 6B). These effects were less pronounced when we used 30 ng/mL and were absent with 3.0 ng/mL pneumolysin or inactive pneumolysins (Figure 2A-B; supplemental Figure 6A-B), consistent with the platelet morphology by electron microscopy (Figure 1F; supplemental Figure 4C-F). We did not observe inhibition in the increase of light transmission induced by pneumolysin after adding 1.16 mM RGDS peptide. RGDS is a potent inhibitor of platelet aggregation (supplemental Figure 7). This indicates that the change in observed light transmission is caused by lysis of platelets rather than by aggregation. RGDS alone did not interfere with pore formation of pneumolysin in cell membranes (supplemental Figure 5A lower left wells). Furthermore, we found that platelets responded only to TRAP-6 either in the presence of very low concentrations of pneumolysin or inactive pneumolysins (Figure 2A-B; supplemental Figure 6A-B).
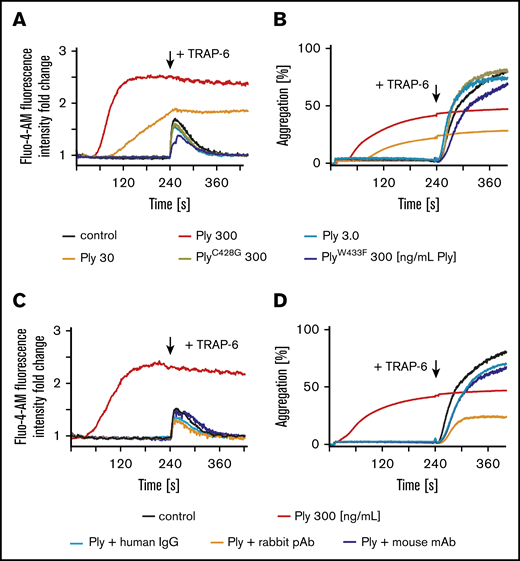
Loss of platelet function due to pneumolysin is prevented by immunoglobulins. (A) Prior to pneumolysin treatment intracellular Ca2+ of washed platelets was labeled with Fluo-4-AM for 30 minutes. After incubation with pneumolysin, the kinetics of Ca2+ release was measured and values are given as fold change compared with NaCl control. Different concentrations of pneumolysin (Ply) are color coded: 300 ng/mL (red); 30 ng/mL (orange); 3.0 ng/mL (light blue). PneumolysinC428G without lytic activity (brown) pneumolysinW433F with ∼10% lytic activity (blue) did not cause Ca2+ release. (B) Platelet aggregation is typically directly proportional to an increase in light transmission. Only pneumolysin 300 ng/mL (red) and 30 ng/mL (orange) induced an increase in light transmission, but platelets where no longer responsive to 20 µM TRAP-6. Light transmission did not change by addition of buffer, pneumolysin 3 ng/mL, or the mutant pneumolysins, but platelets were still responsive to 20 µM TRAP-6. (C-D) Polyvalent human immunoglobulin (human IgG (Privigen); 1 mg/mL; green), polyclonal rabbit anti-pneumolysin (10 µg/mL; orange) and a monoclonal mouse anti-pneumolysin antibody (7.5 µg/mL; blue) prevented the effects of pneumolysin (300 ng/mL; red) in calcium influx (C) and platelet aggregation (D). In the presence of these immunoglobulins platelets became again responsive to 20 µM TRAP-6 (to enable comparison with the experiments without immunoglobulins, the data are shown here, although they are presented in the text at the end of “Results”).
Loss of platelet function due to pneumolysin is prevented by immunoglobulins. (A) Prior to pneumolysin treatment intracellular Ca2+ of washed platelets was labeled with Fluo-4-AM for 30 minutes. After incubation with pneumolysin, the kinetics of Ca2+ release was measured and values are given as fold change compared with NaCl control. Different concentrations of pneumolysin (Ply) are color coded: 300 ng/mL (red); 30 ng/mL (orange); 3.0 ng/mL (light blue). PneumolysinC428G without lytic activity (brown) pneumolysinW433F with ∼10% lytic activity (blue) did not cause Ca2+ release. (B) Platelet aggregation is typically directly proportional to an increase in light transmission. Only pneumolysin 300 ng/mL (red) and 30 ng/mL (orange) induced an increase in light transmission, but platelets where no longer responsive to 20 µM TRAP-6. Light transmission did not change by addition of buffer, pneumolysin 3 ng/mL, or the mutant pneumolysins, but platelets were still responsive to 20 µM TRAP-6. (C-D) Polyvalent human immunoglobulin (human IgG (Privigen); 1 mg/mL; green), polyclonal rabbit anti-pneumolysin (10 µg/mL; orange) and a monoclonal mouse anti-pneumolysin antibody (7.5 µg/mL; blue) prevented the effects of pneumolysin (300 ng/mL; red) in calcium influx (C) and platelet aggregation (D). In the presence of these immunoglobulins platelets became again responsive to 20 µM TRAP-6 (to enable comparison with the experiments without immunoglobulins, the data are shown here, although they are presented in the text at the end of “Results”).
CD62P staining of platelets results from labeling intracellular CD62P
Because the pore formation and the functional experiments in the presence of pneumolysin suggested nonfunctional platelets, we asked why CD62P was upregulated on the platelet membrane, which typically requires platelet activation. By fluorescence microscopy, we visualized that pneumolysin treatment did not result in CD62P surface expression, but that intracellular CD62P was stained (Figure 3; supplemental Figure 8). Staining for CD62P in nonpermeabilized platelets increased with the concentration of pneumolysin (3, 5, 20, 30, 50, 100, 300 ng/mL). At very high pneumolysin concentrations of ≥100 ng/mL, platelets are completely destroyed and both CD62P and α-tubulin (supplemental Figure 9) immunofluorescence staining patterns appear outside in some of the platelets. This potentially results from the breakdown of the fragile platelet plasma membrane during preparation of slides for microscopy by cytospin and during coverslip mounting, which results in appearance of a “squeezing-out” effect of the labeled intracellular proteins from the damaged platelets. Figure 3B shows the cross-sectional fluorescence signal intensity (nonsaturated grayscale median values) for immunofluorescence localization of CD62P in individual platelets (from ≥20 single platelets). The most likely explanation is that pneumolysin-induced pores allowed for antibody penetration into platelets resulting in staining of intracellular CD62P. This is also shown by the control with Triton X-100, which induces pore formation in the platelet membrane. As additional control, we also immunostained for α-tubulin, a strictly intracellular cytoskeletal protein. Staining patterns (supplemental Figure 9A) and cross-sectional fluorescence signal intensities (supplemental Figure 9B; nonsaturated grayscale median values) were similar to the ones obtained for CD62P staining.
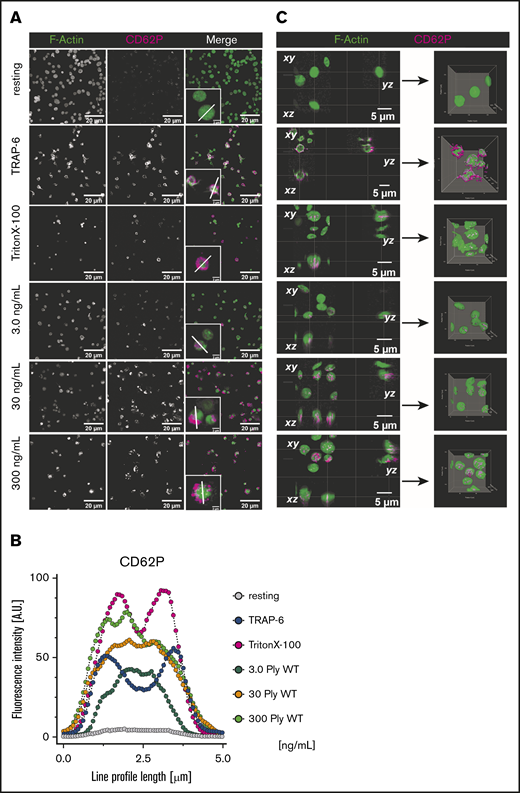
Fluorescence microscopy of pneumolysin-treated platelets. (A) Pneumolysin-treated platelets were stained for F-actin (green) and CD62P (magenta). Platelets were not permeabilized, with the exception of the Triton X-100 control. Insets, Single platelets at higher magnification and the line used for measuring fluorescence intensities shown in panel B. In the presence of 3.0 and 30 ng/mL pneumolysin, intracellular staining of CD62P and α-tubulin become visible. At 200 ng/mL pneumolysin, vesicles staining strongly for pneumolysin surround the platelets (compare Figure 1F). (B) Staining pattern of CD62P throughout single cells treated with pneumolysin was quantified to distinguish between cytoplasmic and only surface-associated CD62P staining. The pattern indicates that CD62P is stained intracellularly and not extracellularly. The different concentrations of pneumolysin used are color coded: 3.0 ng/mL (blue), 30 ng/mL (orange), 300 ng/mL (green). (C) Orthogonal views of confocal Z-stacks and 3D isosurface rendering of pneumolysin-treated platelets stained for F-actin (green) and CD62P (magenta). It shows distinct intracellular accumulation of anti-CD62P antibody in platelets treated with different concentration of pneumolysin and membrane permeabilization with Triton X-100 and surface expression of CD62P upon TRAP-6 stimulation.
Fluorescence microscopy of pneumolysin-treated platelets. (A) Pneumolysin-treated platelets were stained for F-actin (green) and CD62P (magenta). Platelets were not permeabilized, with the exception of the Triton X-100 control. Insets, Single platelets at higher magnification and the line used for measuring fluorescence intensities shown in panel B. In the presence of 3.0 and 30 ng/mL pneumolysin, intracellular staining of CD62P and α-tubulin become visible. At 200 ng/mL pneumolysin, vesicles staining strongly for pneumolysin surround the platelets (compare Figure 1F). (B) Staining pattern of CD62P throughout single cells treated with pneumolysin was quantified to distinguish between cytoplasmic and only surface-associated CD62P staining. The pattern indicates that CD62P is stained intracellularly and not extracellularly. The different concentrations of pneumolysin used are color coded: 3.0 ng/mL (blue), 30 ng/mL (orange), 300 ng/mL (green). (C) Orthogonal views of confocal Z-stacks and 3D isosurface rendering of pneumolysin-treated platelets stained for F-actin (green) and CD62P (magenta). It shows distinct intracellular accumulation of anti-CD62P antibody in platelets treated with different concentration of pneumolysin and membrane permeabilization with Triton X-100 and surface expression of CD62P upon TRAP-6 stimulation.
Thus, we explain the increase in CD62P staining by damaged platelets rather than by platelet activation.
Pneumolysin renders platelets nonfunctional
Consistently, platelets were no longer viable in the presence of 300 ng/mL pneumolysin, whereas 30 ng/mL pneumolysin showed an intermediate phenotype, and 3.0 ng/mL had no effect on viability as measured by an increase of relative luminescence units resulting from intracellular processing of a luminescent substrate. PBS and Triton X-100 were used as controls (Figure 4A; supplemental Figure 10).
![Pneumolysin induces platelet death. (A) Kinetics of platelet viability. PBS was used as viability control and Triton X-100 to induce platelet death. Pneumolysin in increasing concentrations induced platelet death measured by reduced substrate turnover. (B) Platelet viability was maintained in the presence of polyvalent human immunoglobulin (human Ig [Privigen]; 1 mg/mL), polyclonal rabbit anti-pneumolysin (rabbit pAb; 10 µg/mL), or a monoclonal mouse anti-pneumolysin antibody (mouse mAb; 7.5 µg/mL) despite a high concentration of pneumolysin (300 ng/mL). (C) Thrombus formation on collagen in a flow chamber in the absence of pneumolysin was monitored by image acquisition at an interval of 10 seconds by fluorescence microscopy at a shear stress of 1000 s−1. In the presence of pneumolysin, thrombus formation was impaired. (D) Thrombus formation in the presence of pneumolysin was restored by polyvalent immunoglobulin. Human Ig (Privigen) alone had no effect on thrombus formation (supplemental Figure 10). (E) Quantification of the percentage of surface area covered over time by thrombi in the presence of pneumolysin in different concentrations or nonactive pneumolysin mutants. Different concentrations of pneumolysin (Ply) are color coded: 300 ng/mL (red); 30 ng/mL (orange); 3.0 ng/mL (green). PneumolysinC428G without cytolytic activity (brown); pneumolysinW433F with ∼10% cytolytic activity (blue). (F) Quantification of the effect of polyvalent immunoglobulin (human Ig (Privigen); 1 mg/mL) on restoring thrombus formation in the presence of pneumolysin (300 ng/mL). RLU, relative luminescence unit.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/24/10.1182_bloodadvances.2020002372/2/m_advancesadv2020002372f4.png?Expires=1769128141&Signature=P4CO5HrKUl7dEqkgaYBIZb~9ITUngTRlvkWPrhIo2mx2uGfrMu5bOHdXsKUNEUAqNuzB2OxVuIMTq2vssQjYP-V4Mp4zvgWUsId81j6PLIpmuyle-BmF5GePX7UVeZaCv75GXpj0ZxkM-9PRf6gZVBHhJMt0OYNi9WZddn1PvyuooXYFneJuEKC2olD5cgwcCfq4lrvoJu0ErKpQu0GIYEZjFPlCcsmN-9i8VFHZdYRHts-5V~Ocj2ZMU~s76LMORSdr-nKNcC6aWAUQ4KnMtG0ecvrcVcU-mGrXoAbVn8s1W29aEEY7GYaYHlNKsxoTevDwF1WyP1pYNFN74x1A6Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pneumolysin induces platelet death. (A) Kinetics of platelet viability. PBS was used as viability control and Triton X-100 to induce platelet death. Pneumolysin in increasing concentrations induced platelet death measured by reduced substrate turnover. (B) Platelet viability was maintained in the presence of polyvalent human immunoglobulin (human Ig [Privigen]; 1 mg/mL), polyclonal rabbit anti-pneumolysin (rabbit pAb; 10 µg/mL), or a monoclonal mouse anti-pneumolysin antibody (mouse mAb; 7.5 µg/mL) despite a high concentration of pneumolysin (300 ng/mL). (C) Thrombus formation on collagen in a flow chamber in the absence of pneumolysin was monitored by image acquisition at an interval of 10 seconds by fluorescence microscopy at a shear stress of 1000 s−1. In the presence of pneumolysin, thrombus formation was impaired. (D) Thrombus formation in the presence of pneumolysin was restored by polyvalent immunoglobulin. Human Ig (Privigen) alone had no effect on thrombus formation (supplemental Figure 10). (E) Quantification of the percentage of surface area covered over time by thrombi in the presence of pneumolysin in different concentrations or nonactive pneumolysin mutants. Different concentrations of pneumolysin (Ply) are color coded: 300 ng/mL (red); 30 ng/mL (orange); 3.0 ng/mL (green). PneumolysinC428G without cytolytic activity (brown); pneumolysinW433F with ∼10% cytolytic activity (blue). (F) Quantification of the effect of polyvalent immunoglobulin (human Ig (Privigen); 1 mg/mL) on restoring thrombus formation in the presence of pneumolysin (300 ng/mL). RLU, relative luminescence unit.
Pneumolysin induces platelet death. (A) Kinetics of platelet viability. PBS was used as viability control and Triton X-100 to induce platelet death. Pneumolysin in increasing concentrations induced platelet death measured by reduced substrate turnover. (B) Platelet viability was maintained in the presence of polyvalent human immunoglobulin (human Ig [Privigen]; 1 mg/mL), polyclonal rabbit anti-pneumolysin (rabbit pAb; 10 µg/mL), or a monoclonal mouse anti-pneumolysin antibody (mouse mAb; 7.5 µg/mL) despite a high concentration of pneumolysin (300 ng/mL). (C) Thrombus formation on collagen in a flow chamber in the absence of pneumolysin was monitored by image acquisition at an interval of 10 seconds by fluorescence microscopy at a shear stress of 1000 s−1. In the presence of pneumolysin, thrombus formation was impaired. (D) Thrombus formation in the presence of pneumolysin was restored by polyvalent immunoglobulin. Human Ig (Privigen) alone had no effect on thrombus formation (supplemental Figure 10). (E) Quantification of the percentage of surface area covered over time by thrombi in the presence of pneumolysin in different concentrations or nonactive pneumolysin mutants. Different concentrations of pneumolysin (Ply) are color coded: 300 ng/mL (red); 30 ng/mL (orange); 3.0 ng/mL (green). PneumolysinC428G without cytolytic activity (brown); pneumolysinW433F with ∼10% cytolytic activity (blue). (F) Quantification of the effect of polyvalent immunoglobulin (human Ig (Privigen); 1 mg/mL) on restoring thrombus formation in the presence of pneumolysin (300 ng/mL). RLU, relative luminescence unit.
We also demonstrate that platelets in the presence of pneumolysin dose-dependently lose their capability to form thrombi, when whole blood was flown over collagen at arterial shear (1000 s−1) (Figure 4C; supplemental Figure 11). Figure 4D shows the area covered by thrombi in the presence of different concentrations of pneumolysin and inactive pneumolysin mutants. Wild-type but not the inactive pneumolysins inhibited thrombus formation.
One of the major functions of platelets is to seal gaps in the endothelium8 evoked by acute infection.19,20 Pneumolysin inhibits platelet function and thereby compromises their sealing function of the endothelium, increasing the risk of bleeding and fluid extravasation into the interstitial compartment.21,22 We show compromising of this important platelet function using Boyden chamber experiments. Platelets sealed the membrane pores of the Boyden chamber thereby preventing diffusion of fluorescently labeled BSA to the lower chamber. In the presence of pneumolysin, platelets no longer inhibited BSA diffusion (Figure 5).
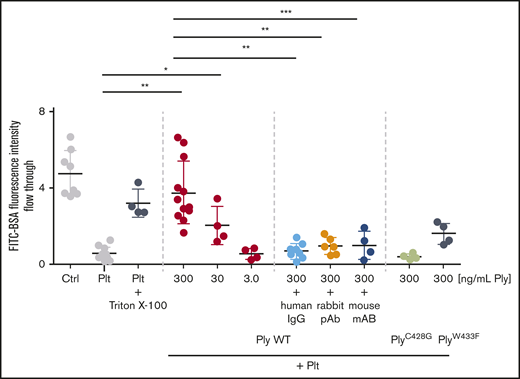
Sealing of Transwell membranes by platelets is impaired by pneumolysin. Platelets seal Transwell membranes with 3-µm pores. This was impaired in the presence of pneumolysin but not the pneumolysin mutants as indicated by the flow through of BSA-FITC to the lower chamber measured by fluorescence intensity (to enable comparison with the experiments without immunoglobulins, the data are shown here, although they are presented in the text at the end of “Results”). *P < .05; **P < .01; ***P < .001.
Sealing of Transwell membranes by platelets is impaired by pneumolysin. Platelets seal Transwell membranes with 3-µm pores. This was impaired in the presence of pneumolysin but not the pneumolysin mutants as indicated by the flow through of BSA-FITC to the lower chamber measured by fluorescence intensity (to enable comparison with the experiments without immunoglobulins, the data are shown here, although they are presented in the text at the end of “Results”). *P < .05; **P < .01; ***P < .001.
Antibodies and polyvalent immunoglobulins inhibit the effects of pneumolysin on platelets in vitro
We then aimed to rescue platelet function in the presence of active pneumolysin by using antibodies neutralizing its cytolytic activity. A monoclonal mouse antibody (7.5 µg/mL), polyclonal rabbit antibodies (10 µg/mL), or a pharmaceutical human IgG preparation (1 mg/mL) completely restored the platelet phenotype and function.
For all impaired functions described in detail herein, we show that: namely, the CD62P expression response to TRAP-6 was restored (Figure 1C; supplemental Figure 12); integrin activation again occurred as measured by PAC-1 binding (supplemental Figure 13); calcium release no longer occurred (Figure 2C; supplemental Figure 14); no pseudoplatelet aggregation (= lysis) was observed (Figure 2D; supplemental Figure 14); thrombus formation in whole blood was again comparable to the normal control (Figure 4D,F; supplemental Figure 11); and platelet viability was rescued, no longer differing from the buffer control (Figure 4B; supplemental Figure 9). Finally, antibodies and immunoglobulins neutralize pneumolysin and platelets remain functional to seal membrane pores of the Boyden chamber (Figure 5).
A polyvalent immunoglobulin preparation was associated with a nominally higher platelet count and nominally reduced mortality in patients with S pneumoniae–induced severe community-acquired pneumonia
In addition to mAbs and pharmaceutical human IgG (IgG-enriched Privigen), the IgM/IgA-enriched immunoglobulin preparation trimodulin is able to effectively neutralize lysis of red cells by pneumolysin (Figure 6A). To translate our findings into clinical practice, we took advantage of an earlier performed phase 2 trial (CIGMA study) in 160 patients with severe community-acquired pneumonia requiring invasive mechanical ventilation. Patients were treated in addition to standard of care with trimodulin (182.6 mg/kg for 5 days) or placebo.17 Infections with S pneumoniae were confirmed in 15 patients in the trimodulin group and in 18 patients in the placebo group. Platelet counts were higher (544 vs 361 platelets per nanoliter on day 14; Figure 6B) and mortality was nominally lower in the trimodulin group (2 of 15 [13.3%] vs 7 of 18 [38.9%]; Figure 6C). A limitation of this study was the small numbers of patients and therefore these preliminary observations warrant further investigation in larger clinical trials.
![Neutralizaton of pneumolysin by trimodulin and relevance for severe community-acquired pneumonia patients. (A) In vitro pneumolysin neutralization assay measuring free hemoglobin as marker for cell lysis, using erythrocytes and different concentrations of trimodulin as indicated. Shown are results from 3 repeated measurements (mean plus or minus standard deviation.). (B) Platelet counts of patients in the CIGMA study with confirmed S pneumoniae infection were obtained before (pre), during (days 2-5), and after (days 6, 7, and 14) treatment with trimodulin or placebo. Pretreatment values were obtained from n = 15 (trimodulin group) and n = 18 (placebo group) and day 14 values obtained from n = 11 (trimodulin group, 1 missing value) and n = 12 patients (placebo group), respectively. (C) The 28-day mortality rate in patients with severe, confirmed S pneumoniae lung infection (n = 15 in trimodulin group, n = 18 in placebo group) was nominally lower in the trimodulin group compared with the placebo group (2 of 15 [13.3%] vs 7 of 18 [38.9%]). Due to small patient numbers, no statistical analysis has been performed. NC, negative control (PBS); PC, positive control (1% Triton X-100).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/24/10.1182_bloodadvances.2020002372/2/m_advancesadv2020002372f6.png?Expires=1769128141&Signature=HlIgl0PWc7XUguiWwJDbCrq5FME9Fg-Fov6dI09742MgPhJkZ2b5fuayDyKc4BbV6w58xTzZ3jQNid5yENeUk~R7rgkrgNBsVPZ0UY2gZa8uTvw8tMB7wQzZ18dF6UWepXVTFYw89xoj6Ar0OOWp-kbZwKdJi8HNtDtjKbV4zAb~oUXZ2Zla1UDJHSNySmpj6aqTLzvjaID9i1VpLFSO15V2YHlzG2~b5s-DujLwjO495CX5BvEUSC-jMyghzrJqdne10fRKW~apc6SPlKtZdCQKNhN03BasjLwS6YZ0X~5s6p68GUF1unws9fnet8fx1fDGt8InEb6NqEkq~soktA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Neutralizaton of pneumolysin by trimodulin and relevance for severe community-acquired pneumonia patients. (A) In vitro pneumolysin neutralization assay measuring free hemoglobin as marker for cell lysis, using erythrocytes and different concentrations of trimodulin as indicated. Shown are results from 3 repeated measurements (mean plus or minus standard deviation.). (B) Platelet counts of patients in the CIGMA study with confirmed S pneumoniae infection were obtained before (pre), during (days 2-5), and after (days 6, 7, and 14) treatment with trimodulin or placebo. Pretreatment values were obtained from n = 15 (trimodulin group) and n = 18 (placebo group) and day 14 values obtained from n = 11 (trimodulin group, 1 missing value) and n = 12 patients (placebo group), respectively. (C) The 28-day mortality rate in patients with severe, confirmed S pneumoniae lung infection (n = 15 in trimodulin group, n = 18 in placebo group) was nominally lower in the trimodulin group compared with the placebo group (2 of 15 [13.3%] vs 7 of 18 [38.9%]). Due to small patient numbers, no statistical analysis has been performed. NC, negative control (PBS); PC, positive control (1% Triton X-100).
Neutralizaton of pneumolysin by trimodulin and relevance for severe community-acquired pneumonia patients. (A) In vitro pneumolysin neutralization assay measuring free hemoglobin as marker for cell lysis, using erythrocytes and different concentrations of trimodulin as indicated. Shown are results from 3 repeated measurements (mean plus or minus standard deviation.). (B) Platelet counts of patients in the CIGMA study with confirmed S pneumoniae infection were obtained before (pre), during (days 2-5), and after (days 6, 7, and 14) treatment with trimodulin or placebo. Pretreatment values were obtained from n = 15 (trimodulin group) and n = 18 (placebo group) and day 14 values obtained from n = 11 (trimodulin group, 1 missing value) and n = 12 patients (placebo group), respectively. (C) The 28-day mortality rate in patients with severe, confirmed S pneumoniae lung infection (n = 15 in trimodulin group, n = 18 in placebo group) was nominally lower in the trimodulin group compared with the placebo group (2 of 15 [13.3%] vs 7 of 18 [38.9%]). Due to small patient numbers, no statistical analysis has been performed. NC, negative control (PBS); PC, positive control (1% Triton X-100).
Discussion
In this study, we assessed the interaction of pneumococci and the toxin pneumolysin with platelets. Taken together, our data lead us to the following conclusions: pneumococci induce pores in the platelet membranes by secretion of pneumolysin. This renders platelets nonfunctional and inhibits platelet-thrombus formation in whole blood. Earlier reports indicating platelet activation by pneumolysin18 are most likely caused by the artifact of anti-CD62P antibody diffusion through pores in the platelet membrane, which then stain intracellularly the activation marker CD62P. Local in vivo concentrations of pneumolysin in the lung are difficult to determine in the flowing blood obtained from patient veins, as the blood has to circulate from the lung through the entire arterial system and capillaries before sampling. Therefore, best estimates on local in vivo concentrations are likely obtained from pneumolysin concentrations in the cerebral fluid obtained from patients with pneumococcal meningitis. Here, pneumolysin concentrations reached up to 30 µg/mL, depending on the report.4,6,7 As we did already observe major damage of platelets in a concentration of 0.03 µg/mL (30 ng/mL), the concentrations, which we have used for our in vitro experiments, are very likely within a clinically relevant range. Importantly, platelet damage depends on the incubation time: the shorter the incubation time, the higher the pneumolysin concentrations needed to damage platelets. In vivo pneumococci infections will last for hours or days and likely rather low concentrations of pneumolysin can induce cell damage in the microenvironment.
We tried to exclude potential artifacts by showing that the increase in light transmission aggregometry was not inhibited by RGDS, performing experiments using washed platelets, platelets in plasma, or platelets in whole blood. All experiments showed consistent results. In this regard, an interesting question is why the many red cells in whole blood do not protect platelets from binding pneumolysin. The concentration of 30 ng/mL pneumolysin at which we observed impairment of thrombus formation in whole blood was the same concentration at which we observed inhibition of platelet function, increased staining of CD62P, or impaired sealing of holes in the Boyden chamber in the absence of red cells. We would have expected that the many red cells outnumbering platelets at least 10 times would quench the effect of pneumolysin on platelets.
It is known that Ca2+ is required to protect the cell membranes from damage by pneumolysin.18 We therefore added Ca2+ in all experiments, but the ones in which we measured calcium release from platelets, because in the latter setting calcium in the buffer would cause artifacts.
Our results are likely clinically relevant. Here, we show that antibodies targeting pneumolysin can inhibit lysis and the loss of platelet function. With pharmaceutical immunoglobulin preparations approved for human use, a ready-to-use intervention is available to interfere with the platelet-damaging effect of pneumococci in patients with acute pneumonia. This may reduce the severity of acute respiratory distress syndrome in these patients. The standard dose of pharmaceutical human IgG is 1 g/kg body weight at 2 consecutive days. Pharmacokinetic studies of pharmaceutical human IgG given in this dose in patients with autoimmune disease show an increase in IgG even 2 weeks after infusion of 7.8 g/L (±5.6 g/L).23
This concept is supported by a post hoc analysis of the CIGMA trial. Although patient numbers with confirmed S pneumoniae infections in this trial are small (15 in the trimodulin vs 18 in the placebo group), the recovery of platelet numbers indicates a potential in vivo effect of immunoglobulins in S pneumoniae–induced pneumonia. However, confirmation of such clinical effects requires larger prospective randomized trials. In such a trial especially, the impact of trimodulin on mortality should be further assessed because we did see a nominally lower mortality in the trimodulin-treated group. In addition, larger patient numbers may also allow us to show the effect of pharmaceutical immunoglobulins at low platelet count levels. In the post hoc analysis, a significant difference between groups was only seen when platelet counts had already recovered to normal levels (544 000 vs 361 000 platelets per microliter). Trimodulin consisting of ∼56% IgG, 23% IgM, and 21% IgA is dosed with a total dose of 0.9 g/kg body weight. Pharmacokinetic analysis from phase 1 testing in 6 healthy volunteers dosed with the same protocol as used in the CIGMA trial demonstrate a maximum plasma concentration of 6 g/L IgG, 1.7 g/L IgM, and 1.5 g/L IgA (S.W. and J.S., Biotest, unpublished data) and therefore plasma levels of IgG achieved by the standard treatment are above the pharmaceutical human IgG concentrations tested in our in vitro experiments.
Which additional role the IgM and IgA components might play in the context of pneumococcal pneumonia is not clear. Possibly, pneumolysin is neutralized by these secretory immunoglobulins already at the primary site of infection in the alveoli before causing larger platelet damage and entering the bloodstream. Previous studies with pharmaceutical human IgG and another IgM/IgA-enriched immunoglobulin preparation showed similar effects on rapid replenishment of platelets in patients and animal models with severe infections.24-26 Interestingly, platelet numbers in these publications were replenished faster in patients or animals when treated with IgM/IgA-enriched preparations or pharmaceutical human IgG compared with controls.25,26
Although our in vitro and in vivo findings require analyses in larger randomized trials, our findings provide a rationale for targeting pneumolysin by use of polyvalent immunoglobulin preparations in severe community-acquired pneumococcal pneumonia to counteract the risk of these patients to become ventilation dependent.
Requests for data may be e-mailed to the corresponding author, Andreas Greinacher.
Acknowledgments
The authors thank Marcus Gutscher, Dennis Riehl, and Katharina Heim for trimodulin assay development, and Alexander Staus for biostatistical analyses of the CIGMA trial at Biotest AG. They are further grateful to Katharina Passvogel, Ina Schleicher, Kristine Sievert-Giermann, and Gerhard Burchhardt for technical support.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG [German Research Foundation] grant number 374031971–TRR 240).
Authorship
Contribution: K.J. performed flow cytometry and cell-viability experiments, contributed to electron microscopy, evaluated the data, prepared the figures and wrote the manuscript; S. Handtke performed platelet-function studies, evaluated the data, prepared figures, and edited the manuscript; R.P. designed and performed flow chamber experiments and platelet confocal microscopy, evaluated the data, prepared figures, and edited the manuscript; S.W., C.H., and J.S. contributed the data on trimodulin and CIGMA studies and edited the manuscript; G.N. and M. Witzenrath contributed to the conceptual design of the study and edited the manuscript; T.P.K. contributed to the flow cytometry experiments and preparation of electron microscopy samples, designed experiments, and edited the manuscript; J.W. contributed to flow cytometry experiments, platelet-function studies, managed healthy donors, and edited the manuscript; M.R. performed the electron microscopy, prepared the figures, and edited the manuscript; A.F.A. and M. Wolff performed the pneumococci-platelet interaction studies and edited the manuscript; S. Hammerschmidt and A.G. designed the project, were responsible for the funding of the project, supervised the project, evaluated the data, and wrote and edited the manuscript; and all authors reviewed the final version of the manuscript
Conflict-of-interest disclosure: A.G. reports grants and nonfinancial support from Aspen, Boehringer Ingelheim, Merck Sharp & Dohme (MSD), Bristol Myers Squibb (BMS), Bayer Healthcare, and Instrumentation Laboratory; personal fees from Aspen, MSD, Macopharma, BMS, Chromatec, and Instrumentation Laboratory; and nonfinancial support from Portola, Ergomed, and Biokit, outside of the submitted work. Charité (G.N. and M. Witzenrath) receives funding for research from Biotest AG. C.H., S.W., and J.S. are employees of Biotest AG. The remaining authors declare no competing financial interests.
Correspondence: Andreas Greinacher, Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, Sauerbruchstr, 17487 Greifswald, Germany; e-mail: andreas.greinacher@med.uni-greifswald.de; and Sven Hammerschmidt, Department of Molecular Genetics and Infection Biology, Interfaculty Institute of Genetics and Functional Genomics, Center for Functional Genomcis of Microbes, Universität Greifswald, Felix-Hausdorff-Str 8, D-17487 Greifswald, Germany; e-mail: sven.hammerschmidt@uni-greifswald.de.

