

Key Points
The Gardos effect drives adhesion molecule activation and vesiculation.
Vesiculation-driven GPC loss activates Lu/BCAM and CD44.

Abstract
Senescence of erythrocytes is characterized by a series of changes that precede their removal from the circulation, including loss of red cell hydration, membrane shedding, loss of deformability, phosphatidyl serine exposure, reduced membrane sialic acid content, and adhesion molecule activation. Little is known about the mechanisms that initiate these changes nor is it known whether they are interrelated. In this study, we show that Ca2+-dependent K+ efflux (the Gardos effect) drives erythrocyte senescence. We found that increased intracellular Ca2+ activates the Gardos channel, leading to shedding of glycophorin-C (GPC)–containing vesicles. This results in a loss of erythrocyte deformability but also in a marked loss of membrane sialic acid content. We found that GPC-derived sialic acid residues suppress activity of both Lutheran/basal cell adhesion molecule (Lu/BCAM) and CD44 by the formation of a complex on the erythrocyte membrane, and Gardos channel–mediated shedding of GPC results in Lu/BCAM and CD44 activation. This phenomenon was observed as erythrocytes aged and on erythrocytes that were otherwise prone to clearance from the circulation, such as sickle erythrocytes, erythrocytes stored for transfusion, or artificially dehydrated erythrocytes. These novel findings provide a unifying concept on erythrocyte senescence in health and disease through initiation of the Gardos effect.
Introduction
The mechanisms that regulate daily erythrocyte sequestration and clearance in the spleen are far from being well understood. However, various processes have been described that precede erythrocyte sequestration. These include vesiculation, and consequently loss of deformability, phosphatidylserine exposure, membrane desialylation, adenosine triphosphate depletion, and adhesion molecule activation.1-5 Some of these are cause-and-effect processes, and others are seemingly unrelated. One of the best described phenomena that relate to erythrocyte senescence during the erythrocyte lifespan is the Gardos effect.5-7 Various studies put forward that as erythrocytes age, adenosine triphosphate depletion and proteolysis of outward rectifying Ca2+ pumps cause intracellular Ca2+ levels to rise.8 As a consequence, the Ca2+-dependent potassium/K+-efflux channel, known as the Gardos channel, is activated, leading to erythrocyte dehydration.9 This phenomenon is observed as erythrocytes age, in sickle cell disease, and in other primary red blood cell disorders that render erythrocytes prone to sequestration in the spleen.6,10,11 The latter is mediated by the endothelial sieve-like fenestrae of the splenic sinuses that ill-deformable erythrocytes are unable to pass.12-14 Thus, Gardos channel–induced dehydration functions to mechanically trap aged, nondeformable erythrocytes that have lost the ability to maintain intracellular homeostasis. The notion that aged erythrocytes, malaria-infected erythrocytes,14,15 sickle erythrocytes,16,17 heat-hardened erythrocytes,18 erythrocytes stored for transfusion, or erythrocytes that are otherwise less deformable,5,6,19 are prone to sequestration in the spleen supports this dogma.
Next to dehydration, membrane desialylation leads to clearance of erythrocytes from the circulation.20 Furthermore, in various hematologic diseases that are characterized by increased erythrocyte turnover and by aged erythrocytes, abnormal activation of Lutheran/basal cell adhesion molecule (Lu/BCAM) and CD44 adhesion molecules is observed.2,3,21-28 Strikingly, loss of membrane sialic acid and erythrocyte adhesion molecule activation are intricately connected. It has been shown that the activity of both Lu/BCAM and CD44 is restricted by membrane sialic acid.2,3 Thus, progressive loss of membrane sialic acid as erythrocytes age29 is directly associated with Lu/BCAM and CD44 activation.2,3 For these reasons, adhesion molecule activation through membrane sialic acid loss has been implicated as a contributor to erythrocyte turnover. Indeed, we were recently able to show that activation of the erythrocyte adhesion molecule is required for erythrocyte turnover.30
Currently, little knowledge is available regarding the physiological mechanisms that underlie membrane desialylation and adhesion molecule activation. It is also unclear what the differential contribution of deformability and adhesion molecule activation is to daily erythrocyte turnover and to erythrocyte turnover under pathological conditions. So, we studied the relationships between the Gardos effect,7 erythrocyte deformability, membrane sialic acid content, and adhesion molecule activation. We show that activation of the Gardos channel drives erythrocyte senescence, and we outline the set of cascading events that connect erythrocyte dehydration to loss of deformability, loss of membrane sialic acid, and adhesion molecule activation.
Methods
Blood samples and isolation of dense and light erythrocytes
Heparinized venous blood was obtained from healthy volunteers after they provided informed consent. Blood studies were approved by the Medical Ethical Committee of Sanquin Research and were performed in accordance with the 2013 Declaration of Helsinki. Erythrocytes were isolated by centrifugation of whole blood at 240g for 15 minutes. Next, plasma and buffy coat were removed, and erythrocytes were washed twice with saline-adenine-glucose-mannitol medium (SAGM medium: 150 mM NaCl, 1.25 mM adenine, 50 mM glucose, 29 mM mannitol [pH 5.6]; Fresenius SE). Washed erythrocytes were then either used for experiments or stored in SAGM for up to 4 weeks. Dense and light erythrocytes were isolated using Percoll (GE Healthcare, Little Chalfont, United Kingdom) density centrifugation. Briefly, isotonic Percoll was prepared by adding 8.1 mL of 10× phosphate-buffered saline (PBS) per 100 mL of Percoll. Next, Percoll buffer (26.3 g/L bovine serum albumin, 132 mM NaCl, 4.6 mM KCl, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) was used to dilute isotonic Percoll to 1.096 g/mL (80%), 1.087 g/mL (71%), 1.083 g/mL (67%), 1.080 g/mL (64%), and 1.060g/mL (40%). Percoll dilutions were stacked in a 15-mL tube, and 2 mL of isolated erythrocytes was added on top and centrifuged at 2100g for 15 minutes at room temperature. Erythrocytes isolated from the fraction denser than 1.096 g/mL Percoll were defined as dense and aged erythrocytes, whereas erythrocytes lighter than 1.080 g/mL Percoll are here defined as light and young erythrocytes.
Generation of GPC KO K562 cells
To generate K562 glycophorin-C (GPC) knockout (KO) cells, we used the CRISPOR Design tool (http://crispor.tefor.net/) to determine the Cas9 target sites present in the coding sequence of GPC. A double-stranded oligonucleotide (ds oligo) (Invitrogen, Carlsbad, CA) was generated from several target sequences, with an additional G 5′ of the target sequence and specific BsmBI overhang. The oligo was then cloned into the BsmBI sites of pLentiCRISPR v2. The constructs were grown in Escherichia coli Stbl3 and sequence verified. Lentiviral particles were generated by transient cotransfection of 293T cells with pLentiCRISPR v2: GPC KO, psPAX2, and pCMV-VSVg. The day after transfection, the cells were put on K562 medium. Virus-containing supernatant was harvested on days 2 and 3 after transfection and filtered through a 0.45 µm filter, and 1 mL was used on 5 × 105 K562 cells on 2 successive days. Transduced K562 cells were selected with 2 μg/mL puromycin (InvivoGen). Expression of GPC was determined by fluorescence-activated cell sorting (FACS), and GPC knockout cells were selected by FACS.
Flow cytometry
Flow cytometric analysis was performed on the LSRII + HTS cell analyzer (BD Biosciences, Franklin Lakes, NJ), and data were analyzed by FACS Diva software (BD Biosciences). Erythrocytes were stained with either 1 µM FLUO-4 (Invitrogen) diluted in dimethyl sulfoxide (DMSO) or with 0.1 µM potassium-binding benzofuran isophthalate acetoxymethyl ester (PBFI) (Invitrogen) diluted in DMSO and supplemented with a previous treatment of 0.4% pluronic with 500 μM propranolol or 10 μM valinomycin (all from Sigma-Aldrich, St. Louis, MO) for 30 minutes at 37°C. Blocking experiments were performed by prestimulating cells with 25 µM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), 10 µM 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole (TRAM-34), 25 µM Calpain 1 inhibitor (A6185), and 1 µg/mL diisopropylfluorophosphate (DFP) (all from Sigma-Aldrich) or 40 µM N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone (Z-VAD-FMK) (R&D Systems, Minneapolis, MN) for 15 minutes at 37°C and then stimulating with propranolol for 30 minutes at 37°C. Vehicle controls were applied for every control condition. α2,3-Linked sialic acid was quantified by flow cytometry using biotinylated Maackia amurensis lectin II (Vector Laboratories, Peterborough, United Kingdom) followed by streptavidin alexa fluor 488 conjugation (Thermo Fisher Scientific, Waltham, MA). Glycophorin-A and GPC expression on erythrocytes was quantified by anti-glycophorin-A (GPA) PE (M1732; Sanquin, Amsterdam, The Netherlands) and anti-GPC (BRIC10 and BRIC4, a kind gift from International Blood Group Reference Laboratory (IBGRL, Bristol, United Kingdom).
Intracellular K+-content measurement, erythrocyte deformability, and surface area
At first, 40 μL of packed erythrocytes were subjected to hemolysis by treating cells with 200 μL demineralized water for 10 minutes on ice. Intracellular K+ content and hemoglobin were measured in the supernatant of hemolyzed erythrocytes by using the Rapidlab 865 blood gas analyzer (Siemens Medical Solution Diagnostics, Erlangen, Germany). Deformability of young and aged erythrocytes was measured using an automated rheoscope and cell analyzer (ARCA) at a shear stress of 30 dyne/cm2 (3 Pa) as described previously.31 Erythrocyte surface area was measured with ARCA under a sheer stress of 10 dyne/cm2 (1 Pa).
Immunoprecipitation and western blot
For immunoprecipitation, 75 µL Protein G Dynabeads (Thermo Fisher Scientific) was washed twice in PBS and incubated with 30 µL of anti-CD44 fluorescein isothiocyanate (Diaclone, Besançon, France) or mouse immunoglobulin G isotype control (Thermo Fisher Scientific) antibody for 1 hour in 300 µL PBS. Then, 2 × 108 erythrocytes were treated with or without neuraminidase (Vibrio cholerae filtrate, Sigma-Aldrich) for 15 minutes at 37°C and lysed in lysis buffer (10 mM Tris, 30 mM NaCl, 0.5% NP40, 10% glycerol, and 1:100 HALT protease inhibitor cocktail) (Thermo Fisher Scientific) and incubated with antibody-coated protein-G beads overnight at 4°C. Afterward, the protein-G–antibody target complex was washed 5 times in lysis buffer supplemented with HALT and subjected to Laemmli sample buffer, run on sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Whatman, Little Chalfont, United Kingdom) for detection.
Flow assays
Erythrocyte adhesion to laminin-α5 and hyaluronic acid (HA) was assessed by coating 0.5 µg laminin-511 (BioLamina, Sundbyberg, Sweden) or 7.5 µg HA (Sigma-Aldrich) diluted in HEPES buffer (132 mM NaCl, 20 mM HEPES, 6 mM KCl, 1 mM MgSO4, 1.2 mM K2HPO4, 1 mM Ca2+ [all from Sigma-Aldrich) per lane through passive adsorption on uncoated IBIDI μ-slide VI0.4 or ibiTreat µ-slide VI0.4 flow chambers (IBIDI). Erythrocytes diluted in HEPES+ medium (HEPES buffer as described above supplemented with 0.5% human serum albumin and 1 mg/mL glucose) were flowed via a syringe pump system over either laminin-α5– or HA-coated microslides at a flowrate of 0.2 dynes/cm2. Adhesion was measured by taking 9 frames with EVOS microscopy (Thermo Fisher Scientific) 15 minutes after injecting erythrocytes to laminin-α5–coated flow chambers. Erythrocyte rolling on HA-coated flow chambers was assessed by taking 9 frames after 10 minutes of adding cells to the flow assay system and quantifying rolling and tethering erythrocytes. Laminin-α5–adherent andHA-rolling erythrocytes were applied by image analysis software Vision4D (Arivis, Rostock, Germany). The adhesion and rolling frequencies were calculated by normalizing the different conditions to the ratio of the baseline control:vehicle sample, which equals 1. Experimental data were analyzed using Graphpad Prism 6 software. Data are presented as mean ± standard deviation, unless otherwise indicated in the figure legends. The data were assumed to follow a normal distribution.
Results
Erythrocyte dehydration is associated with sialic acid loss and adhesion molecule activation
Aged erythrocytes, sickle erythrocytes, and erythrocytes stored for transfusion are characterized by an increased propensity for being removed from the circulation.6,16,32 Senescent erythrocytes are characterized by low intracellular potassium content (Figure 1A), reduced deformability (Figure 1B), and reduced levels of membrane sialic acid (Figure 1C). Furthermore, these erythrocytes express activated Lu/BCAM and CD44 adhesion molecules, as is illustrated by their increased adhesion to laminin-α5 and HA (Figure 1D-E). Erythrocytes isolated from patients with sickle cell anemia were not fractionated; however, they exhibit impaired characteristics similar to those observed in aged erythrocytes (Figure 1 A-E). The observation that aged, stored, and sickle erythrocytes that are prone to removal from the circulation are less deformable, contain reduced membrane sialic acid, and display increased adhesion to laminin-α5 and HA led us to hypothesize that these phenomena are interrelated. To test for this, we treated healthy erythrocytes with vehicle or 10 μM of the K+ ionophore valinomycin, thereby inducing dehydration through potassium efflux (Figure 1F). These cells also displayed a marked decrease in deformability (Figure 1G), contained reduced membrane sialic acid content (Figure 1H), and exhibited increased adhesion and rolling to laminin-α5 and HA, respectively (Figure 1I). These phenomena were also observed when storing erythrocytes for 4 weeks in the SAGM transfusion storage buffer, where a gradual increase in adhesion frequency was noted (Figure 1J-K) Together, these findings suggested a correlation between erythrocyte hydration state and sialic acid loss, simultaneously affecting membrane deformability and adhesion molecule activation.
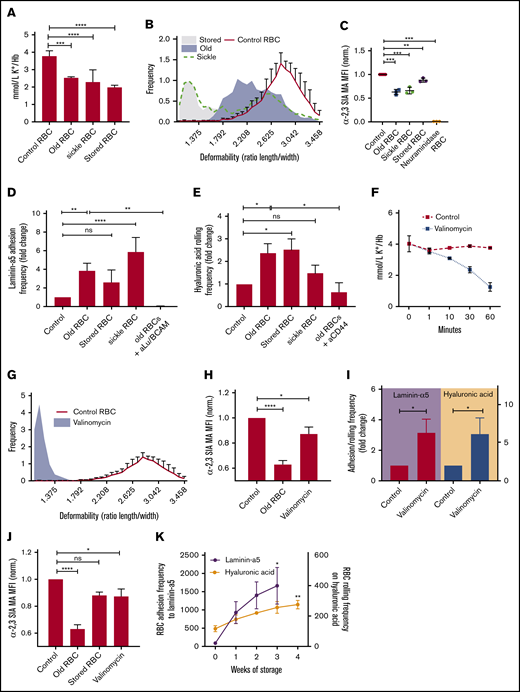
Erythrocyte dehydration is associated with adhesion molecule activation. (A) Intracellular K+ content in aged, sickle cell, and stored erythrocytes was measured using ion-specific electrodes (n = 4-8; 1-way ANOVA). (B) Erythrocyte deformability was determined using the automated rheologic cell analyzer (ARCA). A shear stress of 30 dyne/cm2 was applied to determine deformability (ratio of length (A) to width (B) of the cell (n = 3; 1-way ANOVA). (C) Biotinylated Maackia amurensis (MA) lectin was used to quantify α2,3-linked sialic acid (α2,3-SIA) on aged, stored (in SAGM medium), and sickle erythrocytes; data were normalized (Norm.) to untreated controls (n = 3; 1-way ANOVA). (D) A total of 107 control, aged, stored, and sickle erythrocytes were flowed over laminin-α5 at 0.2 dynes/cm2 and adhesion frequency was assessed by microscopy (n = 3-7; 1-way ANOVA). (E) The same flow experiment was performed on HA. Here, rolling frequency was quantified instead of adhesion (n = 3-7; 1-way ANOVA). (F) Intracellular K+ content in control and valinomycin-treated cells was measured using ion-specific electrodes over 60 minutes. (G) ARCA was performed to assess deformability of erythrocytes treated for 30 minutes with valinomycin (n = 3). (H) Biotinylated M amurensis lectin was used to quantify α2,3-linked sialic acid on erythrocytes exposed for 30 minutes to valinomycin; data were normalized to untreated controls (n = 3-4, 1-way ANOVA). (I) A total of 107 control and valinomycin-treated erythrocytes were flowed over laminin-α5 and HA at 0.2 dynes/cm2 and adhesion frequency was assessed by microscopy. Erythrocytes were treated with valinomycin for 30 minutes because this results in levels of intracellular potassium similar to those in aged, sickle, and stored erythrocytes (panel A) (n = 4-9; Student t test). (J) Flow cytometric quantification of α2,3-linked sialic acid on old, stored, and valinomycin-treated erythrocytes (n = 3-4; 1-way ANOVA). (K) Erythrocytes were stored for up to 4 weeks at 4°C during which adhesion frequency to laminin-α5 and rolling frequency on HA was assessed. Significance compared with T = 0 is indicated (1-way ANOVA). Error bars indicate standard deviation unless otherwise noted. *P < .05; **P < .01; ***P < .001; ****P < .0001. Hb, hemoglobin; MFI, mean fluorescence intensity; ns, not significant; RBC, red blood cell.
Erythrocyte dehydration is associated with adhesion molecule activation. (A) Intracellular K+ content in aged, sickle cell, and stored erythrocytes was measured using ion-specific electrodes (n = 4-8; 1-way ANOVA). (B) Erythrocyte deformability was determined using the automated rheologic cell analyzer (ARCA). A shear stress of 30 dyne/cm2 was applied to determine deformability (ratio of length (A) to width (B) of the cell (n = 3; 1-way ANOVA). (C) Biotinylated Maackia amurensis (MA) lectin was used to quantify α2,3-linked sialic acid (α2,3-SIA) on aged, stored (in SAGM medium), and sickle erythrocytes; data were normalized (Norm.) to untreated controls (n = 3; 1-way ANOVA). (D) A total of 107 control, aged, stored, and sickle erythrocytes were flowed over laminin-α5 at 0.2 dynes/cm2 and adhesion frequency was assessed by microscopy (n = 3-7; 1-way ANOVA). (E) The same flow experiment was performed on HA. Here, rolling frequency was quantified instead of adhesion (n = 3-7; 1-way ANOVA). (F) Intracellular K+ content in control and valinomycin-treated cells was measured using ion-specific electrodes over 60 minutes. (G) ARCA was performed to assess deformability of erythrocytes treated for 30 minutes with valinomycin (n = 3). (H) Biotinylated M amurensis lectin was used to quantify α2,3-linked sialic acid on erythrocytes exposed for 30 minutes to valinomycin; data were normalized to untreated controls (n = 3-4, 1-way ANOVA). (I) A total of 107 control and valinomycin-treated erythrocytes were flowed over laminin-α5 and HA at 0.2 dynes/cm2 and adhesion frequency was assessed by microscopy. Erythrocytes were treated with valinomycin for 30 minutes because this results in levels of intracellular potassium similar to those in aged, sickle, and stored erythrocytes (panel A) (n = 4-9; Student t test). (J) Flow cytometric quantification of α2,3-linked sialic acid on old, stored, and valinomycin-treated erythrocytes (n = 3-4; 1-way ANOVA). (K) Erythrocytes were stored for up to 4 weeks at 4°C during which adhesion frequency to laminin-α5 and rolling frequency on HA was assessed. Significance compared with T = 0 is indicated (1-way ANOVA). Error bars indicate standard deviation unless otherwise noted. *P < .05; **P < .01; ***P < .001; ****P < .0001. Hb, hemoglobin; MFI, mean fluorescence intensity; ns, not significant; RBC, red blood cell.
Gardos channel-driven dehydration regulates deformability and adhesion molecule activation
Erythrocytes dehydrate as they age as a consequence of the inability to maintain low intracellular Ca2+ levels, which is observed during erythrocyte ageing and storage and in sickle cell disease.6,9,33 Increased intracellular Ca2+ concentrations lead to the activation of the Ca2+-dependent K+ efflux channel known as the Gardos channel.6,9 Concomitant uncompensated efflux of K+ is accompanied by a reduction of intracellular H2O, resulting in erythrocyte dehydration, which is also known as the Gardos effect.7,9 Because valinomycin is a crude stimulus that directly induces K+ efflux, we sought a stimulus to induce erythrocyte dehydration through the Gardos effect.5,17 Because propranolol, a β2-adrenergic receptor antagonist, has been described to induce potassium efflux at high concentrations,34 we assessed whether this is achieved through the Gardos effect.7 We found that treatment of erythrocytes with 1 mM propranolol resulted in a transient increase of intracellular Ca2+ content (Figure 2A). This was followed by a gradual decrease of intracellular K+ content (Figure 2B) and induced a stomatocyte-like phenotype (Figure 2B, right panel), suggesting Gardos channel activation. To confirm this, we blocked the Gardos channel with 10 μM TRAM34 or vehicle before propranolol treatment and found normalization of intracellular K+ content (Figure 2C). This shows that propranolol is a potent inducer of the Gardos effect, which we then used to study adhesion molecule activation as a consequence of Gardos channel activation. We determined that propranolol induces loss of erythrocyte deformability (Figure 2D) and membrane sialic acid content (Figure 2E). Furthermore, a significant increase in Lu/BCAM and CD44-mediated binding of erythrocytes to laminin-α5 and HA was observed (Figure 2F). This was dependent on activation of the Gardos effect because blocking the Gardos channel with TRAM34 in the presence of propranolol resulted in normalization of the adhesion molecule activation state (Figure 2F). Together, these data directly link activation of the Gardos channel, that in vivo is associated with erythrocyte dehydration and sequestration,11 to loss of deformability, loss of membrane sialic acid, adhesion molecule activation and thus, to erythrocyte senescence.
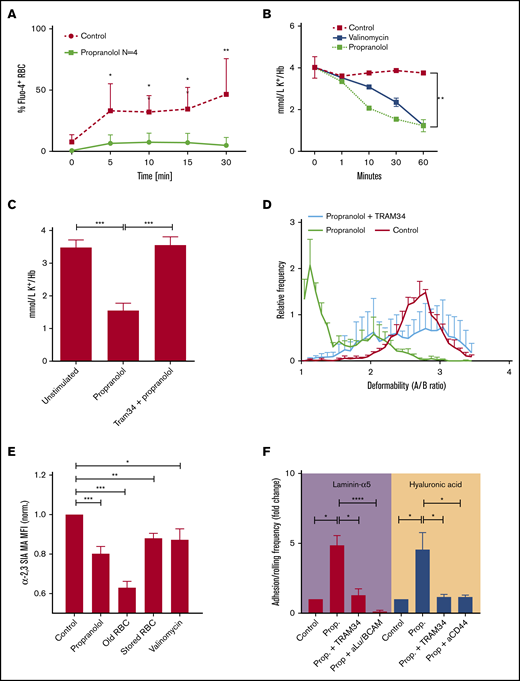
Propranolol induces Gardos channel–mediated erythrocyte dehydration. (A) Cells were stained with Fluo-4 to measure influx of Ca2+ in response to propranolol (Prop.) (n = 4; 1-way ANOVA). (B-C) K+ content in erythrocytes treated with propranolol or propranolol plus TRAM34 (n = 3; Student t test). (D) ARCA was performed to assess deformability of propranolol-treated erythrocytes (n = 3; Student t test). A/B ratio represents length over width ratio of the cells under shear. (E) Biotinylated M amurensis lectin was used to quantify α2,3-linked sialic acid on propranolol, aged, stored, and valinomycin-treated erythrocytes; data were normalized to untreated controls. (F) Effect of control, propranolol, and TRAM34 on adhesion and rolling frequency of erythrocytes to laminin-α5 and HA, respectively (n = 3-9; Student t test). Error bars indicate standard deviation unless otherwise noted. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Propranolol induces Gardos channel–mediated erythrocyte dehydration. (A) Cells were stained with Fluo-4 to measure influx of Ca2+ in response to propranolol (Prop.) (n = 4; 1-way ANOVA). (B-C) K+ content in erythrocytes treated with propranolol or propranolol plus TRAM34 (n = 3; Student t test). (D) ARCA was performed to assess deformability of propranolol-treated erythrocytes (n = 3; Student t test). A/B ratio represents length over width ratio of the cells under shear. (E) Biotinylated M amurensis lectin was used to quantify α2,3-linked sialic acid on propranolol, aged, stored, and valinomycin-treated erythrocytes; data were normalized to untreated controls. (F) Effect of control, propranolol, and TRAM34 on adhesion and rolling frequency of erythrocytes to laminin-α5 and HA, respectively (n = 3-9; Student t test). Error bars indicate standard deviation unless otherwise noted. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Gardos channel–dependent vesiculation results in sialic acid loss and adhesion molecule activation
Next, we aimed to determine the mechanisms by which Gardos channel activation results in membrane desialylation. Endogenous erythrocyte sialidases35,36 were found to be unlikely to be causative for this because Arachis hypogea lectin, a lectin that agglutinates erythrocytes that are exposed to sialidase activity,37 did not react with aged, stored, sickle, or valinomycin- or propranolol-stimulated erythrocytes, in contrast to neuraminidase-treated erythrocytes (Figure 3A). Morphologic analysis and imaging flow cytometry revealed that propranolol treatment resulted in stomatocytosis (Figure 3B), reduction in cell size (Figure 3C), and vesiculation (Figure 3D). Because proteolysis has previously been observed during vesiculation,38 we blocked various proteases during propranolol treatment, aiming to prevent vesiculation and adhesion molecule activation. In contrast to various protease inhibitors such as Z-VAD-FMK and a Calpain I inhibitor, we found that DFP, a serine protease inhibitor, potently prevented vesiculation (Figure 3E) and membrane sialic acid loss (Figure 3F). This was associated with inhibition of adhesion molecule activation (Figure 3G). However, DFP did not seem to directly inhibit vesiculation. Rather, it was found to modulate Gardos channel activity because K+ efflux and morphology remained unaffected (Figure 3H-I) and Ca2+ influx was not inhibited (Figure 3J). These data show that vesiculation that takes place after Gardos channel activation causes membrane sialic acid loss through vesiculation. Furthermore, these results indicate that DFP renders the Gardos channel less sensitive to influx of Ca2+. Thus, Ca2+ influx alone is not enough to affect adhesion molecule activation. Instead, the Gardos effect (ie, erythrocyte dehydration and vesiculation) is required to exert this effect.
![The Gardos effect drives vesiculation-dependent sialic acid loss and adhesion molecule activation. (A) Control, aged, valinomycin-treated (Val.), stored, propranolol-treated (Prop.), sickle, and neuraminidase-treated (N’ase) erythrocytes (1 × 108) were incubated with Arachis hypogea lectin. Exposure of the T-antigen that follows removal of terminal sialic acid residues will allow A hypogea lectin to agglutinate these cells. Neuraminidase treatment was used as a positive control. (B) Micrograph of erythrocytes treated with propranolol. Although a minority of erythrocytes remained round and biconcave, a large fraction of erythrocytes had already assumed stomatocyte-like appearance with shrinking and loss of biconcavity. The width of the exerpt is 50 μm. (C) ARCA quantification of erythrocyte surface area under a sheer stress of 10 dynes/cm2. (D) Imagestream analysis was performed to identify erythrocytes and vesicles in response to propranolol treatment. (E) Shedding of relatively large vesicles (>0.3 µm) in response to propranolol treatment was quantified by flow cytometry. (F) Effect of propranolol and inhibition of serine proteases by DFP on expression of α2,3-linked membrane sialic acid. (G) A total of 107 control, propranolol-treated, and DFP-pretreated erythrocytes were flown over laminin-α5 and HA at 0.2 dynes/cm2, and adhesion frequency was assessed by microscopy. (H) Erythrocytes were stained with the Ca2+ dye Fluo-4 and treated with propranolol or with DFP before treatment with propranolol. Flow cytometry was performed to quantify Ca2+ influx in response to these stimuli (n = 3; 1-way ANOVA). (I) Relative intracellular potassium levels were assessed by potassium-binding benzofuran isophthalate acetoxymethyl ester (PBFI) staining and flow cytometry. PBFI was used instead of ion-specific electrodes because this allowed us to work with fewer erythrocytes, which enabled us to detect the effect of the various protease inhibitors (n = 3-4; 1-way ANOVA). (J) Micrograph of erythrocyte morphology upon treatment with various protease inhibitors and propranolol. The induction of stomatocytosis by propranolol was quantified by flow cytometry based on an increase in side scatter. This was also used to measure the degree of inhibition of stomatocytosis by various protease inhibitors (n = 3; 1-way ANOVA). *P < .05; **P < .01; ***P < .001. FSC, forward scatter; inh, inhibitor; SSC, side scatter; ZVAD, Z-VAD-FMK (N-benzyloxycarbonyl-Val-Ala-Asp[O-Me] fluoromethyl ketone).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/24/10.1182_bloodadvances.2020003077/1/m_advancesadv2020003077f3.png?Expires=1769218390&Signature=jP2HBgzlxAal--16sAAZEO4zWa46yLHYDJUfRJ8Y20a5X1F87xqIt23ZwLD1yJpxJsuLsNuMyFP-IqJwC-cGUYffSS5rSm1t8CZ86Pd5Z9bC92~u2dWrLHx5jY0omhvHWtH08ES~VTGrco~8K5us4LYn7hIAie78KjbrxWuRS0ofYAvpSk2PnOpEZQh~6tUInPdkL7P5g5ZmDpxidmGK~yU-WI4-TvwKJrpPeqPbmXz5YjG0Wu~TPOUXuGRB8izw0PUQwjNTNGjY0BhErwgWnsIQp9DtEYg5d-Pf80oxNlIGEUe1YF6EO0ym88Lw7MsvzSKCo7ZUwGyet99OOj79Mg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The Gardos effect drives vesiculation-dependent sialic acid loss and adhesion molecule activation. (A) Control, aged, valinomycin-treated (Val.), stored, propranolol-treated (Prop.), sickle, and neuraminidase-treated (N’ase) erythrocytes (1 × 108) were incubated with Arachis hypogea lectin. Exposure of the T-antigen that follows removal of terminal sialic acid residues will allow A hypogea lectin to agglutinate these cells. Neuraminidase treatment was used as a positive control. (B) Micrograph of erythrocytes treated with propranolol. Although a minority of erythrocytes remained round and biconcave, a large fraction of erythrocytes had already assumed stomatocyte-like appearance with shrinking and loss of biconcavity. The width of the exerpt is 50 μm. (C) ARCA quantification of erythrocyte surface area under a sheer stress of 10 dynes/cm2. (D) Imagestream analysis was performed to identify erythrocytes and vesicles in response to propranolol treatment. (E) Shedding of relatively large vesicles (>0.3 µm) in response to propranolol treatment was quantified by flow cytometry. (F) Effect of propranolol and inhibition of serine proteases by DFP on expression of α2,3-linked membrane sialic acid. (G) A total of 107 control, propranolol-treated, and DFP-pretreated erythrocytes were flown over laminin-α5 and HA at 0.2 dynes/cm2, and adhesion frequency was assessed by microscopy. (H) Erythrocytes were stained with the Ca2+ dye Fluo-4 and treated with propranolol or with DFP before treatment with propranolol. Flow cytometry was performed to quantify Ca2+ influx in response to these stimuli (n = 3; 1-way ANOVA). (I) Relative intracellular potassium levels were assessed by potassium-binding benzofuran isophthalate acetoxymethyl ester (PBFI) staining and flow cytometry. PBFI was used instead of ion-specific electrodes because this allowed us to work with fewer erythrocytes, which enabled us to detect the effect of the various protease inhibitors (n = 3-4; 1-way ANOVA). (J) Micrograph of erythrocyte morphology upon treatment with various protease inhibitors and propranolol. The induction of stomatocytosis by propranolol was quantified by flow cytometry based on an increase in side scatter. This was also used to measure the degree of inhibition of stomatocytosis by various protease inhibitors (n = 3; 1-way ANOVA). *P < .05; **P < .01; ***P < .001. FSC, forward scatter; inh, inhibitor; SSC, side scatter; ZVAD, Z-VAD-FMK (N-benzyloxycarbonyl-Val-Ala-Asp[O-Me] fluoromethyl ketone).
The Gardos effect drives vesiculation-dependent sialic acid loss and adhesion molecule activation. (A) Control, aged, valinomycin-treated (Val.), stored, propranolol-treated (Prop.), sickle, and neuraminidase-treated (N’ase) erythrocytes (1 × 108) were incubated with Arachis hypogea lectin. Exposure of the T-antigen that follows removal of terminal sialic acid residues will allow A hypogea lectin to agglutinate these cells. Neuraminidase treatment was used as a positive control. (B) Micrograph of erythrocytes treated with propranolol. Although a minority of erythrocytes remained round and biconcave, a large fraction of erythrocytes had already assumed stomatocyte-like appearance with shrinking and loss of biconcavity. The width of the exerpt is 50 μm. (C) ARCA quantification of erythrocyte surface area under a sheer stress of 10 dynes/cm2. (D) Imagestream analysis was performed to identify erythrocytes and vesicles in response to propranolol treatment. (E) Shedding of relatively large vesicles (>0.3 µm) in response to propranolol treatment was quantified by flow cytometry. (F) Effect of propranolol and inhibition of serine proteases by DFP on expression of α2,3-linked membrane sialic acid. (G) A total of 107 control, propranolol-treated, and DFP-pretreated erythrocytes were flown over laminin-α5 and HA at 0.2 dynes/cm2, and adhesion frequency was assessed by microscopy. (H) Erythrocytes were stained with the Ca2+ dye Fluo-4 and treated with propranolol or with DFP before treatment with propranolol. Flow cytometry was performed to quantify Ca2+ influx in response to these stimuli (n = 3; 1-way ANOVA). (I) Relative intracellular potassium levels were assessed by potassium-binding benzofuran isophthalate acetoxymethyl ester (PBFI) staining and flow cytometry. PBFI was used instead of ion-specific electrodes because this allowed us to work with fewer erythrocytes, which enabled us to detect the effect of the various protease inhibitors (n = 3-4; 1-way ANOVA). (J) Micrograph of erythrocyte morphology upon treatment with various protease inhibitors and propranolol. The induction of stomatocytosis by propranolol was quantified by flow cytometry based on an increase in side scatter. This was also used to measure the degree of inhibition of stomatocytosis by various protease inhibitors (n = 3; 1-way ANOVA). *P < .05; **P < .01; ***P < .001. FSC, forward scatter; inh, inhibitor; SSC, side scatter; ZVAD, Z-VAD-FMK (N-benzyloxycarbonyl-Val-Ala-Asp[O-Me] fluoromethyl ketone).
Shedding of GPC+ vesicles induces Lu/BCAM and CD44 activation
Next, we tried to unravel the mechanism by which membrane sialic acid loss through vesiculation results in Lu/BCAM and CD44 activation. Recent studies have shown that Lu/BCAM is inactivated by binding to GPC-derived sialic acid residues on healthy erythrocytes.3 We thus questioned whether GPC expression levels are diminished after dehydration by propranolol and valinomycin, which would explain why Lu/BCAM is activated in response to erythrocyte dehydration. By means of flow cytometry, we found that GPC expression on propranolol- and valinomycin-dehydrated erythrocytes is strongly reduced (Figure 4A) similar to what is observed on aged erythrocytes.3 Interestingly, expression of GPA, a glycophorin that is more abundantly expressed than GPC and also highly glycosylated, was found to be stable during dehydration (Figure 4A), suggesting that protein sorting to vesicles or vesicle budding and concomitant adhesion molecule activation is a specific and regulated process. These findings were confirmed by imaging flow cytometry, showing that propranolol-induced dehydration causes shedding of GPC+ vesicles that are devoid of GPA (Figure 4B). These findings explain why Lu/BCAM is activated during erythrocyte dehydration.
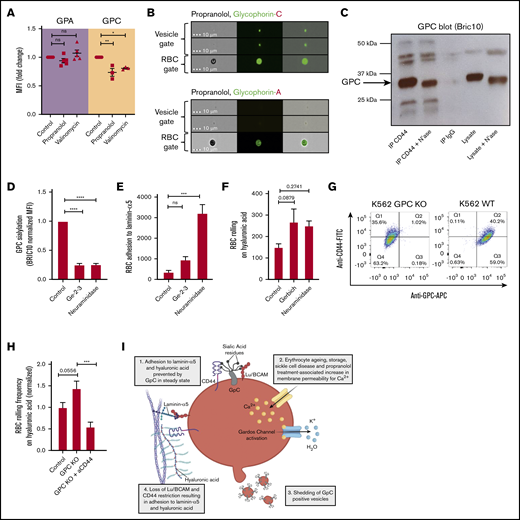
Shedding of GPC+vesicles lift the restriction on Lu/BCAM and CD44 activity. (A) Expression of anti-GPA/GPC on control, propranolol, and valinomycin-treated erythrocytes. MFI was normalized to control cells and expressed as fold change. (B) Imagestream analysis was performed to identify erythrocyte and vesicle populations (see Figure 3B) in response to propranolol treatment. The distribution of GPA and GPC on erythrocytes and their vesicles was determined (lower panel). (C) CD44 immunoprecipitation (IP) was performed on control and neuraminidase-treated erythrocytes and blotted for GPC using BRIC10 monoclonal antibody. Band height difference between control and neuraminidase-treated erythrocytes indicate successful desialylation of GPC. Immunoglobulin G (IgG) control and lysates were taken along as controls. (D) We quantified expression of a sialylated epitope of GPC using BRIC10 on control erythrocytes and on erythrocytes lacking exon 3 of GPC known as the Gerbich (Ge+2–3) phenotype. Neuraminidase-treated erythrocytes were taken along as control (n = 3-7; 1-way ANOVA). (E-F) A total of 107 control, Ge–2–3 and neuraminidase-treated erythrocytes were flowed over laminin-α5 and HA at 0.2 dynes/cm2, and adhesion/rolling frequency was assessed by microscopy (n = 3-5; 1-way ANOVA). (G) Flow cytometry staining of CD44-FITC and GPC-allophycocyanin (APC) on wild-type (WT) K562 and GPC knockout (KO) K562 cells. IgG controls were used for gating. (H) Adhesion of WT and GPC KO K562 cells to HA. (I) Schematic representation (created with Biorender.com) of senescence of RBCs as a consequence of Gardos activation. First, calcium enters the cell, either through a stimulus such as propranolol or as a consequence of transient leakage of calcium such as that observed during erythrocyte ageing. The increased intracellular calcium levels activate the calcium-dependent potassium efflux channel known as the Gardos channel, causing RBC dehydration. In response to dehydration, erythrocytes shed vesicles that contain GPC, causing loss of membrane sialic acid which directly results in Lu/BCAM and CD44 activation.2,3 *P < .05; **P < .01; ***P < .001; ****P < .0001. FITC, fluorescein isothiocyanate.
Shedding of GPC+vesicles lift the restriction on Lu/BCAM and CD44 activity. (A) Expression of anti-GPA/GPC on control, propranolol, and valinomycin-treated erythrocytes. MFI was normalized to control cells and expressed as fold change. (B) Imagestream analysis was performed to identify erythrocyte and vesicle populations (see Figure 3B) in response to propranolol treatment. The distribution of GPA and GPC on erythrocytes and their vesicles was determined (lower panel). (C) CD44 immunoprecipitation (IP) was performed on control and neuraminidase-treated erythrocytes and blotted for GPC using BRIC10 monoclonal antibody. Band height difference between control and neuraminidase-treated erythrocytes indicate successful desialylation of GPC. Immunoglobulin G (IgG) control and lysates were taken along as controls. (D) We quantified expression of a sialylated epitope of GPC using BRIC10 on control erythrocytes and on erythrocytes lacking exon 3 of GPC known as the Gerbich (Ge+2–3) phenotype. Neuraminidase-treated erythrocytes were taken along as control (n = 3-7; 1-way ANOVA). (E-F) A total of 107 control, Ge–2–3 and neuraminidase-treated erythrocytes were flowed over laminin-α5 and HA at 0.2 dynes/cm2, and adhesion/rolling frequency was assessed by microscopy (n = 3-5; 1-way ANOVA). (G) Flow cytometry staining of CD44-FITC and GPC-allophycocyanin (APC) on wild-type (WT) K562 and GPC knockout (KO) K562 cells. IgG controls were used for gating. (H) Adhesion of WT and GPC KO K562 cells to HA. (I) Schematic representation (created with Biorender.com) of senescence of RBCs as a consequence of Gardos activation. First, calcium enters the cell, either through a stimulus such as propranolol or as a consequence of transient leakage of calcium such as that observed during erythrocyte ageing. The increased intracellular calcium levels activate the calcium-dependent potassium efflux channel known as the Gardos channel, causing RBC dehydration. In response to dehydration, erythrocytes shed vesicles that contain GPC, causing loss of membrane sialic acid which directly results in Lu/BCAM and CD44 activation.2,3 *P < .05; **P < .01; ***P < .001; ****P < .0001. FITC, fluorescein isothiocyanate.
As for CD44, various modes of regulating its activation have been described. These include masking of the HA-binding epitope of CD44 by its own terminal sialic acid residues,39 labile disulphide bonds that are reduced in steady state preventing HA recognition,40 and HA-binding states that, to various degrees, may depend on the former.41 Thus, both structural regulation and accessibility of the HA-binding groove are important in binding CD44 to HA. Because we observed CD44 activation in response to shedding of vesicles containing highly sialylated proteins such as GPC, we questioned whether CD44 may (similar to Lu/BCAM) bind GPC in a sialic acid–dependent fashion, thus preventing CD44 from interacting with HA. By using co-immunoprecipitation, we found that CD44 (similar to Lu/BCAM3 ) is in complex with GPC (Figure 4C). However, in contrast to Lu/BCAM,3 this interaction was found to be sialic acid independent because sialic acid stripping of erythrocytes by neuraminidase treatment before co-immunoprecipitation did not abolish the interaction between CD44 and GPC (Figure 4C). Thus, CD44 activation did not seem to derive from abrogation of a sialic acid–dependent interaction with GPC, in contrast to Lu/BCAM.3 Alternatively, we hypothesized that GPC may inhibit CD44 from interacting with HA in a similar manner because CD44-derived terminal sialic acid residues interfere with this interaction through shielding of the HA-recognizing epitope.39 This would explain why CD44 is activated in response to neuraminidase treatment2 and in response to dehydration-induced GPC and sialoglycan shedding.
To test this hypothesis, we isolated erythrocytes from individuals that lack exon 3 of the GPC protein, also known as the Gerbich (Ge–2–3) phenotype. Sialylation of GPC on erythrocytes from these individuals is considerably reduced compared with wild-type erythrocytes. This was determined by staining with BRIC10, a monoclonal antibody that recognizes a sialylated epitope on GPC (Figure 4D). The reduction in GPC sialylation was found to correlate strongly with increased binding of these erythrocytes, not only to laminin-α5 as previously reported,3 but also to HA (Figure 4E-F). This shows that GPC sialylation is important for inhibition of CD44 activation. To prove that GPC inhibits CD44 adhesion to HA, we performed the same flow experiments with undifferentiated CD44+GPC+ K562 cells and CRISPR-Cas9 genetically modified undifferentiated CD44+GPC– K562 cells (Figure 4G). We found that K562 cells lacking GPC were interacting significantly more frequent with HA compared with wild-type erythrocytes (Figure 4H). Together, these data show that erythrocyte dehydration through the Gardos effect7 is a master regulator associated with loss of membrane deformability and loss of GPC that functions to restrict Lu/BCAM and CD44 activation (Figure 4I).
Discussion
In this article, we describe the cascade of events that result from initiation of the Gardos effect,7 mapped the concomitant changes that erythrocytes undergo, and defined their senescence. Similar to results from studies of eryptosis, our results indicate that increased intracellular Ca2+ levels induce membrane vesiculation, reduced deformability, cell shrinkage, and the activation of the Gardos channel.42,43 However, we observed that calpains are not involved in vesicle shedding or in adhesion molecule activation upon treatment with propranololin in contrast to classic eryptosis.44,45 We found that initiation of the Gardos effect7 results in activation of adhesion molecules and loss of deformability, which was also observed in aged, sickle, and stored or otherwise dehydrated erythrocytes. Adhesion molecule activation and loss of erythrocyte deformability are 2 factors that are associated with erythrocyte turnover.2,3,10,16,46
Gardos “channelopathy” has already been described in the context of sickle cell disease in which patients were treated with Senicapoc, an inhibitor of the Gardos channel, to prevent vaso-occlusive crises.17 Although treatment with Senicapoc did not prevent vaso-occlusion, it did reduce reticulocytosis and increased hemoglobin concentrations.17 These findings indicated that the lifespan of sickle erythrocytes was increased by preventing erythrocyte dehydration. Similarly, Fermo et al11 reported Gardos channel hyperactivation in 2 patients who suffered from hemolytic anemia as a result of a mutation in the Gardos channel gene KCNN4. Here we show that adhesion molecules are gradually activated as erythrocytes dehydrate and become less deformable. Therefore, it is extremely difficult to differentiate between the contributions of the 2 processes of erythrocyte turnover. Thus, a stimulus or situation in which only dehydration or only adhesion molecule activation is affected is of prime interest in determining their differential contribution to erythrocyte turnover. We anticipated that the In(Lu) phenotype is such a condition, because In(Lu) erythrocytes have greatly reduced expression of Lu/BCAM and CD44. However, the level of expression of these receptors on the erythrocytes of different In(Lu) individuals varies. Furthermore, because proteins such as Lu/BCAM are linked to the spectrin network, membrane instability is observed in the In(Lu) phenotype, leading to mild acanthocytosis,47 which may also affect mechanical retention.
Another interesting deficiency is the Leach phenotype (Ge–2–3–4), which results in the absence of GPC in erythrocytes48 and is associated with mild hemolytic anemia. However, GPC similarly links to the spectrin network, inducing elliptocytosis in Leach phenotype erythrocytes,48 again also affecting red blood cell deformability. One very compelling hereditary condition in this context is Southeast Asian ovalocytosis (SAO). SAO is caused by a heterozygous partial deletion of band 3,49 which leads to alterations in erythrocyte hydration state50 and causes marked rigidification of erythrocytes.51 Surprisingly, these individuals are clinically asymptomatic with no signs of anemia.52 These individuals, along with the majority of elliptocytosis patients, are examples in which marked loss of erythrocyte deformability is not associated with their massive sequestration, arguing in favor of the hypothesis that next to mechanical retention, other factors such as adhesion molecule activation as a result of Ca2+ influx and Gardos channel activation, may contribute to erythrocyte turnover. Thus, it would be of interest to study adhesion molecule activation on these erythrocytes.
Next to Lu/BCAM and CD44, erythrocytes express various other adhesion molecules such as ICAM-4 and CD147, but their function on mature erythrocytes is still unknown. Similarly, not much is known about the distribution of the ligands of these adhesion molecules, such as laminin-α5 and HA, in organs such as the spleen and liver. Addressing these issues may further elucidate the function of these adhesion molecules and reveal why they are specifically activated on aged, stored, and sickle erythrocytes that are prone to being removed from the circulation.
Membrane vesiculation has been linked to avoidance of complement-mediated destruction indicated by the detection of terminal complement components C5b-9 on Ca2+-induced vesicles.53 Interestingly, complement protein C5b-7 interacts with sialic acid residues54 suggesting that highly sialylated vesicles potentially initiate the reported formation of the C5b-9 complex on these vesicles.
On the basis of the findings presented here, it is clear that hemolytic anemias that arise as a consequence of increased erythrocyte Ca2+ permeability and subsequent Gardos channel activation may be especially suitable for treatment with Gardos channel blockers such as Senicapoc. Treatment of sickle cell disease with Senicapoc has already been shown to result in increased erythrocyte lifespan. And patients with hereditary xerocytosis who were suffering from increased Ca2+ permeability as a result of PIEZO1 and KCNN4 gain-of-function mutations were responsive to Senicapoc.55-57 These patients may be especially responsive to treatment with Gardos channel blockers to prevent or dampen the observed hemolytic anemia.
Requests for data sharing should be e-mailed to the corresponding author, Robin van Bruggen (r.vanbruggen@sanquin.nl).
Acknowledgment
This work was supported by a grant from the Landsteiner Foundation for Blood Transfusion Research (LSBR1412) (T.R.L.K.).
Authorship
Contributors: T.R.L.K. designed the experimental setup, performed experiments, designed figures, and wrote the manuscript; B.M.B., J.J.D., M.V., I.M.S., F.A.I., and P.J.J.H.V. performed the experiments; P.C.L. provided rare blood group samples; T.W.K. and R.v.Z. supervised the experiments; and R.v.B. supervised the experiments and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robin van Bruggen, Sanquin Research and Laboratory Services, Department of Blood Cell Research, Plesmanlaan 125, 1066CX Amsterdam, The Netherlands; e-mail: r.vanbruggen@sanquin.nl.

