

Key Points
Myeloid neoplasm (MPN) clones can evolve from acute myeloid leukemia to gain dominance with isocitrate dehydrogenase inhibition.
Pro-differentiation agents such as ivosidenib can unmask MPN sequelae.

Introduction
Acute myeloid leukemia (AML) can either evolve from antecedent myeloid neoplasms (MPN) or arise de novo.1 Typically, AML disease progression occurs in stepwise fashion as transformed clones increasingly acquire genetic and epigenetic complexity, culminating in sometimes aggressive and fatal chemoresistant leukemias. Targeted therapies, such as isocitrate dehydrogenase (IDH) inhibitors, have transformed the treatment landscape for AML.2,3 Incorporation of these agents into the standard of care has resulted in a shift away from genotoxic therapies and toward differentiation therapies, which has consequently altered the selection pressures that favor the outgrowth of different clones. Here, we report a case of a patient diagnosed with complex karyotype/IDH1-mutant AML whose persistent disease after multiple regimens was treated with ivosidenib. Notably, the patient exhibited signs of robust count recovery on ivosidenib, which coincided with the emergence of a dominant JAK2V617F-mutant clone, progressing to overt polycythemia requiring cytoreduction with hydroxyurea and therapeutic phlebotomy. Droplet digital polymerase chain reaction (PCR) revealed a small detectable JAK2V617F clone that was below the threshold of detection by bulk-targeted sequencing, and single-cell sequencing studies demonstrated that this IDH1R132C co-mutant clone gained dominance after ivosidenib. This case highlights the potential for IDH inhibitors to create selection pressures that promote the outgrowth of minor MPN clones and cause the clinical sequelae of polycythemia in an AML patient without antecedent MPN.
Case description
A 35-year-old man was diagnosed with AML with myelodysplasia-related changes, associated with complex karyotype and RUNX1D198N, DNMT3AR882H, and IDH1R132C mutations and underwent induction chemotherapy with mitoxantrone, etoposide, and cytarabine (Table 1). He achieved a morphologic leukemia-free state, which precluded him from enrolling on a clinical trial, and was instead treated with decitabine for 2 cycles and found to have rising blasts in the marrow. He then enrolled on a clinical trial and was treated with an investigational IDH1 inhibitor FT-2102 for 13 cycles with persistent but stable disease. The patient underwent a repeat bone marrow biopsy that revealed persistent AML with a similar molecular profile except for a newly discovered JAK2V617F mutation (Figure 1A-D; Table 1).
Timeline of mutations and cytogenetic abnormalities
| Date | Therapy | Hb | Blast % | JAK2 p.V617F | DNMT3A p.R882H | IDH1 p.R132C | RUNX1 p.D171N | NRAS p.G12D | NRAS p.Q61L | ATM p.Q355H | Cytogenetics |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | MEC induction | 7.5 | 27 | Neg | 35% | 30% | 46% | Neg | Neg | Neg | 49,XY,+Y,+8,+21[20]; FISH +8 and +21 (no % information, outside report) |
| 2 mo | Decitabine (2 cycles) | ||||||||||
| 5 mo | IDH1 inhibitor FT-2102 (13 cycles) | ||||||||||
| 15 mo | ND | 36 | Neg | 42.99% | 57.39% | 58.93% | Neg | Neg | Neg | 49,XY,+Y,+8,+21[19]/50,idem,+8[1]; FISH +8 and +21 (85.3%) | |
| 17 mo | 12.8 | 49 | 13.54% | 41.67% | 56.23% | 57.26% | Neg | Neg | Neg | 49,XY,+Y,+8,+21[19]/46,XY[1]; FISH: +8/+21 (84%) | |
| 18 mo | High-dose cytarabine reinduction | ||||||||||
| 19 mo | Azacitidine venetoclax (1 cycle) | 7.5 | 55 | 1.26% | 14.61% | 22.05% | 22.38% | Neg | Neg | Neg | FISH: +8 and +21 (42.3%) |
| 21 mo | Ivosidenib | 8.3 | 76 | 0.92% | 37.07% | 35.24% | 52.39% | Neg | Neg | Neg | FISH: +8 and +21 (47%) |
| 23 mo | 18.2 | 17 | 81.34% | 44.44% | 46.51% | 62.27% | Neg | Neg | 11.40% | 49,XY,+Y,+8,+21[20]; FISH: +8 and +21 (95.7%) | |
| 25 mo | 11.7 | 89 | 92.35% | 46.52% | 43.55% | 59.53% | Neg | 24.52% | 15.09% | 49,XY,+Y,+8,+21[19]/46,XY[1] | |
| 26 mo | Decitabine ruxolitinib (4 cycles) | ||||||||||
| 30 mo | Low-dose cytarabine venetoclax (1 cycle) | 12.6 | 80 | 92.22% | 43.68% | 45.52% | 64.24% | 4.39% | Neg | 43.57% | 49,XY,+Y,+8,+21[20]; FISH: +Y (73%) |
| Date | Therapy | Hb | Blast % | JAK2 p.V617F | DNMT3A p.R882H | IDH1 p.R132C | RUNX1 p.D171N | NRAS p.G12D | NRAS p.Q61L | ATM p.Q355H | Cytogenetics |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | MEC induction | 7.5 | 27 | Neg | 35% | 30% | 46% | Neg | Neg | Neg | 49,XY,+Y,+8,+21[20]; FISH +8 and +21 (no % information, outside report) |
| 2 mo | Decitabine (2 cycles) | ||||||||||
| 5 mo | IDH1 inhibitor FT-2102 (13 cycles) | ||||||||||
| 15 mo | ND | 36 | Neg | 42.99% | 57.39% | 58.93% | Neg | Neg | Neg | 49,XY,+Y,+8,+21[19]/50,idem,+8[1]; FISH +8 and +21 (85.3%) | |
| 17 mo | 12.8 | 49 | 13.54% | 41.67% | 56.23% | 57.26% | Neg | Neg | Neg | 49,XY,+Y,+8,+21[19]/46,XY[1]; FISH: +8/+21 (84%) | |
| 18 mo | High-dose cytarabine reinduction | ||||||||||
| 19 mo | Azacitidine venetoclax (1 cycle) | 7.5 | 55 | 1.26% | 14.61% | 22.05% | 22.38% | Neg | Neg | Neg | FISH: +8 and +21 (42.3%) |
| 21 mo | Ivosidenib | 8.3 | 76 | 0.92% | 37.07% | 35.24% | 52.39% | Neg | Neg | Neg | FISH: +8 and +21 (47%) |
| 23 mo | 18.2 | 17 | 81.34% | 44.44% | 46.51% | 62.27% | Neg | Neg | 11.40% | 49,XY,+Y,+8,+21[20]; FISH: +8 and +21 (95.7%) | |
| 25 mo | 11.7 | 89 | 92.35% | 46.52% | 43.55% | 59.53% | Neg | 24.52% | 15.09% | 49,XY,+Y,+8,+21[19]/46,XY[1] | |
| 26 mo | Decitabine ruxolitinib (4 cycles) | ||||||||||
| 30 mo | Low-dose cytarabine venetoclax (1 cycle) | 12.6 | 80 | 92.22% | 43.68% | 45.52% | 64.24% | 4.39% | Neg | 43.57% | 49,XY,+Y,+8,+21[20]; FISH: +Y (73%) |
Hb, hemoglobin; MEC, mitoxantrone, etoposide, and cytarabine; ND, no data; neg, negative.
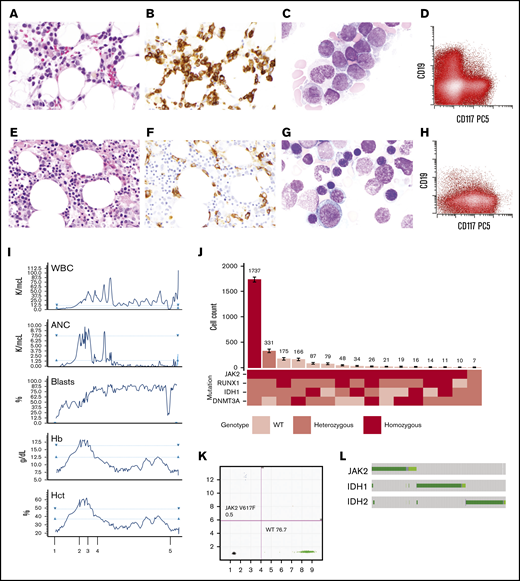
Evolution and emergence of a dominant JAK2V617Fand IDH1R132Cco-mutant clone induced by ivosidenib. Bone marrow morphology with hemoxylin and eosin stain (A), CD34 immunostain (B), Wright-Giemsa stain of bone marrow aspirate (C), and immunophenotype of the leukemic blasts (D) before initiating IDH inhibitor treatment. Bone marrow morphology with hemoxylin and eosin stain (E), CD34 immunostain (F), Wright-Giemsa stain of bone marrow aspirate (G), and immunophenotype of the leukemic blasts (H) following treatment with ivosidenib. (I) Timeline of complete blood count results in relation to therapy: 1, start of ivosidenib; 2 and 3, therapeutic phlebotomy; 4, decitabine/ruxolitinib; and 5, low-dose cytarabine/venetoclax. (J) Clonal architecture analysis using single-cell sequencing. Bar plot (top) depicts the number of cells identified with a given genotype (bottom) ranked by decreasing frequency of the clone. Cell counts for each clone is depicted with error bars derived from random resampling analysis. (K) Droplet digital PCR results on the marrow before IDH inhibitor treatment. (L) Illustration of JAK2, IDH1, and IDH2 mutational exclusivity patterns in AML from cBioportal. ANC, absolute neutrophil count; Hct, hematocrit; PCS, phycoerythrin-Cy5; WBC, white blood cell; WT, wild-type.
Evolution and emergence of a dominant JAK2V617Fand IDH1R132Cco-mutant clone induced by ivosidenib. Bone marrow morphology with hemoxylin and eosin stain (A), CD34 immunostain (B), Wright-Giemsa stain of bone marrow aspirate (C), and immunophenotype of the leukemic blasts (D) before initiating IDH inhibitor treatment. Bone marrow morphology with hemoxylin and eosin stain (E), CD34 immunostain (F), Wright-Giemsa stain of bone marrow aspirate (G), and immunophenotype of the leukemic blasts (H) following treatment with ivosidenib. (I) Timeline of complete blood count results in relation to therapy: 1, start of ivosidenib; 2 and 3, therapeutic phlebotomy; 4, decitabine/ruxolitinib; and 5, low-dose cytarabine/venetoclax. (J) Clonal architecture analysis using single-cell sequencing. Bar plot (top) depicts the number of cells identified with a given genotype (bottom) ranked by decreasing frequency of the clone. Cell counts for each clone is depicted with error bars derived from random resampling analysis. (K) Droplet digital PCR results on the marrow before IDH inhibitor treatment. (L) Illustration of JAK2, IDH1, and IDH2 mutational exclusivity patterns in AML from cBioportal. ANC, absolute neutrophil count; Hct, hematocrit; PCS, phycoerythrin-Cy5; WBC, white blood cell; WT, wild-type.
The patient underwent reinduction with high-dose cytarabine but did not achieve a remission. He received one cycle of azacitidine and venetoclax without response and then began treatment with the approved IDH1 inhibitor ivosidenib. After 2 months on ivosidenib, his pancytopenia resolved and he developed leukocytosis with neutrophilia, polycythemia (hematocrit >60%), and increased circulating blasts. A bone marrow biopsy revealed persistent AML with decreased blasts (17%) since initiation of ivosidenib but with marked erythroid hyperplasia and persistent IDH1 and JAK2 mutations with a markedly increased JAK2V617F VAF of 81.6% (Figure 1E-H; Table 1). Ivosidenib was held periodically for polycythemia and the patient required therapeutic phlebotomy as well as hydroxyurea for cytoreduction (Figure 1I). The patient then began treatment with 4 cycles of decitabine and ruxolitinib followed by low-dose cytarabine and venetoclax for persistent disease. He was eventually hospitalized for neutropenic sepsis and died shortly thereafter, approximately 1 month after the initiation of low-dose cytarabine/venetoclax.
Methods
Single-cell DNA sequencing and analysis was performed using the Mission Bio Tapestri platform, as previously described.4 Bulk sequencing was performed using Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets-heme-targeted sequencing, as previously described.5 Bone marrow aspirates were used for chromosome and fluorescence in situ hybridization (FISH) analysis following a standard protocol. Locus-specific probes for gain of chromosome 8 and 21 and Y chromosome (Abbott Molecular, Des Plaines, IL) were used. Chromosome and FISH abnormalities were reported following the international system of human chromosome nomenclature (ISCN 2016).
The specimens analyzed by sequencing and PCR-based approaches in this manuscript were obtained from the Memorial Sloan-Kettering Cancer Center institutional review board-approved biospecimen bank.
Results and discussion
Targeted sequencing of serial bone marrow aspirates demonstrated RUNX1, IDH1, and DNMT3A mutations throughout the clinical course. Interestingly, the JAK2V617F mutation was undetectable at presentation but was first detected with a variant allele frequency (VAF) of 13.5% by bulk sequencing after treatment with an investigational IDH1 inhibitor, FT-2102. Two months after the patient was started on a second IDH1 inhibitor, ivosidenib, the mutant JAK2 VAF rose further to 81%, consistent with loss of heterozygosity at the JAK2 locus. Additionally, the patient exhibited signs of differentiation with a rise in both neutrophils and hemoglobin, to the extent of meeting all major World Health Organization criteria for polycythemia vera despite persistent AML, which required periodic phlebotomy and hydroxyurea for cytoreduction.
To deconvolute the clonal architecture of this patient’s AML/MPN at the time he developed polycythemia on ivosidenib therapy, we performed single-cell DNA sequencing on peripheral blood mononuclear cells, which identified multiple clones with mutations involving JAK2, RUNX1, IDH1, and DNMT3A (Figure 1J). Homozygous JAK2V617F mutations were present in almost every clone except for 1. The most dominant clone accounted for 62.5% of the patient’s mononuclear cells and was homozygous for JAK2V617F and co-mutant for heterozygous mutations in RUNX1D198N, DNMT3AR882H, and IDH1R132C. Notably, the majority of the unique clones shared JAK2 and IDH1 mutations.
We next sought to determine if the JAK2V617F mutation was detectable earlier than originally identified in the patient’s disease course via standard resolution next-generation targeted sequencing. To that end, DNA was extracted from viably cryopreserved mononuclear cells and subjected to droplet digital PCR. This assay confirmed that very low frequencies of droplets harboring the JAK2-mutant allele were present and detectable with an imputed VAF of 0.6%, which is below the detection threshold of bulk next-generation targeted sequencing (Figure 1K).
These molecular assays at 2 different points in this patient’s AML course provide insightful molecular snapshots that track how a minor JAK2V617F clone that was initially below the limit of detection by bulk sequencing became dominant after differentiation therapy and subsequently evolved further with various patterns of co-mutation with DNMT3A, IDH1, and RUNX1. This clonal dominance coincided with clinical manifestations of MPN with polycythemia and evidence of myeloid differentiation with neutrophilia. Previous work from McKenney et al6 evaluated the treatment response of JAK2/IDH co-mutant MPN mouse models and primary MPN patient samples to IDH inhibition and revealed that combination therapy with the JAK2 inhibitor ruxolitinib and enasidenib normalized aberrant stem and progenitor compartments and reduced disease burden relative to IDH or JAK inhibitor monotherapy. Interestingly, enasidenib monotherapy in mouse models caused increased hematocrit and induced erythroid differentiation in MPN patient samples grown in methylcellulose assays. Although it remains possible that the outgrowth of the JAK2-mutant clone could have occurred irrespective of IDH inhibition,7 these preclinical data are consistent with the hypothesis that IDH inhibition creates selection pressures that favor the differentiation and outgrowth of JAK2/IDH co-mutant clones, similar to what was observed in this patient. However, a small retrospective analysis of MPN patients harboring IDH2 mutations treated with enasidenib revealed that IDH inhibitors can provide some treatment responses in this patient population.8 Moreover, these data are consistent with recent studies showing that enasidenib induces erythroid differentiation independent of its on-target effect on mutant IDH2,9 raising the possibility that JAK2-mutant cells primed to differentiate toward an erythroid cell fate may be further enhanced with IDH inhibition.
To investigate the frequency of co-mutation between JAK2 and IDH1/2, we queried our cBioPortal database across all AML and found that, although patients with concurrent mutations do exist, it is relatively rare (Figure 1L). However, MPN patients with both JAK2 and IDH1/2 mutations, particularly with primary myelofibrosis, have inferior overall survival and leukemia-free survival10 and represent an area in need of further investigation, including credentialing of therapeutic vulnerabilities in this subset of patients. In summary, we report a rare case of MPN that became unmasked in the setting of IDH1 inhibitor differentiation therapy for relapsed AML. This case illustrates how selection pressures exerted by targeted therapies can alter the course of disease through perturbations in clonal dynamics.
Sequencing data will be shared upon request to the corresponding authors, Sheng F. Cai (cais1@mskcc.org) or Ross L. Levine (leviner@mskcc.org).
Acknowledgments
This work was supported by a Young Investigator Award from the Conquer Cancer Foundation (S.F.C.), Career Development Program Fellow Awards from the Leukemia & Lymphoma Society (S.F.C. and L.A.M.), a Young Investigator Award from The Truth 365 (S.F.C.), a Momentum Award from the Mark Foundation for Cancer Research (S.F.C.), and an ASH Scholar Award (S.F.C.). This research was also funded in part through a Cancer Center Support grant from the National Institutes of Health National Cancer Institute (P30 CA008748).
Authorship
Contribution: W.X., T.M., S.E.S., M.S.T., R.L.L., and S.F.C. wrote the manuscript; V.D., H.S.T., and N.L.D. processed samples for sequencing and droplet digital PCR; L.A.M. and R.L.B. performed single-cell DNA sequencing with computational analysis; M.Z. and Y.Z. analyzed cytogenetics and molecular sequencing data; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: W.X. has received research support from Stemline Therapeutics. S.F.C. has consulted for and holds equity interest in Imago Biosciences. R.L.L. serves on the supervisory board of Qiagen and is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis; he receives research support from and has consulted for Celgene and Roche; receives research support from Prelude Therapeutics; has consulted for Novartis and Gilead; and has received honoraria from Lilly and Amgen for invited lectures. L.A.M. has received speaking honoraria from Mission Bio, Inc. M.S.T. receives research funding from AbbVie, Cellerant, Orsenix, ADC Therapeutics, Biosight, receives royalties from UpToDate, and serves on the advisory boards of AbbVie, BioLineRx, Daiichi-Sankyo, Orsenix, KAHR, Rigel, Nohla, Delta-Fly Pharma, and Tetraphase. The remaining authors declare no competing financial interests.
Correspondence: Sheng F. Cai, Leukemia Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: cais1@mskcc.org; or Ross L. Levine, Leukemia Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: leviner@mskcc.org.

