

Key Points
Late-onset and long-lasting autoimmune neutropenia are different entities compared with primary autoimmune neutropenia.
Autoimmune neutropenia presenting at >3 years of age, with no tendency to resolution, would deserve extensive immunologic investigations.

Abstract
Primary autoimmune neutropenia (pAN) is typified by onset in early infancy and a mild/moderate phenotype that resolves within 3 years of diagnosis. In contrast, secondary AN is classically an adult disease associated with malignancy, autoimmunity, immunodeficiency, viral infection, or drugs. This study describes a cohort of 79 children from the Italian Registry who, although resembling pAN, did not fully match the criteria for pAN because neutropenia either appeared after age 5 years (LO-Np) or lasted longer than 3 years (LL-Np). These 2 categories compared with classical pAN showed a far inferior rate of resolution (P < .001), lower severity of neutropenia (P = .03), leukopenia (P < .001), lymphopenia (P < .001) with low B+ (P = .001), increased need of granulocyte colony-stimulating factor (P = .04), and increased frequency of autoimmunity over the disease course (P < .001). A paired comparison between LO-Np and LL-Np suggested that LO-Np had a lower rate of resolution (P < .001) and lower white blood cell (P < .001) and lymphocyte (P < .001) values, higher occurrence of apthae (P = .008), and a stronger association with autoimmune diseases/markers (P = .001) than LL-Np, thus suggesting a more pronounced autoimmune signature for LO-Np. A next-generation sequencing panel applied in a small subgroup of LO-Np and LL-Np patients identified variants related to immune dysregulations. Overall, these findings indicate that there are important differences among pAN LL-Np and LO-Np. Forms rising after 3 years of age, with low tendency to resolution, require tight monitoring and extensive immune investigations aimed to early identify underlying immunologic disease.
Introduction
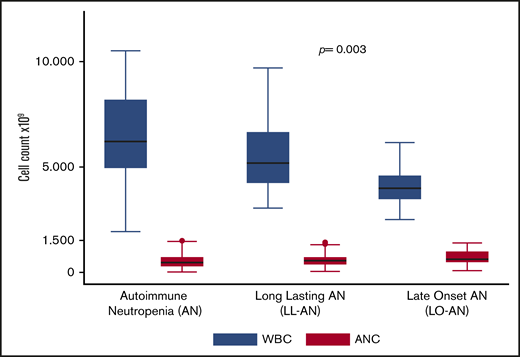
WBCs and absolute neutrophils count: differences between AN, LL-Np, and LO-Np.
Patients and methods
AN was defined by the presence of an ANC (confirmed on ≥3 samples) below the threshold for age, lasting longer than 3 months, and associated with positive indirect anti–neutrophil antibody testing (antibodies found in the patient’s serum) as detected through granulocyte indirect immunofluorescence test (GIFT). GIFT was centralized only in 1 laboratory and was performed ≥3 times for all the patients with the aim to overcome the well-known lack of sensitivity.1,6,7
Two categories of autoimmune neutropenia patients were eligible for the study. The first included subjects diagnosed at age ≥5 years up to 18 years who were defined as late-onset neutropenia (LO Np). The second included children diagnosed at <3 years of age, whose neutropenia lasted >3 years, and who were called long-lasting neutropenia (LL Np).
Neutropenia resolution was defined when ANC were stably >1.5 × 109/Lin ≥3 samples within 3 months.
Patients with AN were also excluded if AN was associated at diagnosis with autoimmune markers or disorders (systemic autoimmune diseases, primary immunodeficiencies, common variable immunodeficiencies defined according to the European Society of Immunodeficiency) criteria (with the exception of isolated immunoglobulin A deficiency) or with concomitant additional cytopenias.8,9 Also excluded were all patients with neutropenia associated with drug assumption, neoplasms, or that occurred after HSCT or in whom GIFT tested negative for ≥3 times. Criteria for treatment with granulocyte colony-stimulating factor (G-CSF) were those of the Italian Guidelines.10
Demographic, clinical, biochemical, immunologic, and genetic data of patients were extracted from the database of the Italian Neutropenia Registry (INR).
Data were collected according to Helsinki declaration of principles. Each patient or his/her guardian signed an informed consent form. Molecular analysis was performed with a next-generation sequencing (NGS) through the HaloPlex Target Enrichment System, including 315 genes involved in hematologic disorders, immunodeficiencies, immune dysregulation, and inflammatory and bone marrow failure syndromes. Variants were confirmed by Sanger sequencing. Clinical and immunologic data were compared with those from a group of patients included in the INR who entirely fulfilled the diagnostic criteria of pAN that were as follows: onset in early infancy at <3 years of age, positive GIFT indirect anti–neutrophil antibody test, mild/moderate infection load, no association with autoimmune markers or disease, and resolution of neutropenia within 36 months since diagnosis.
Statistical analysis
Descriptive analysis has been reported as absolute frequency for qualitative data and according to median, range, and standard deviation for quantitative data. The χ2 test was used for frequency distribution (and Fisher’s test for expected frequency <5). The Mann-Whitney U test was used for nonparametric comparison, and the Kruskall-Wallis test was used for comparison between groups. Bonferroni's adjustment was used to compare the ratings for each group. Values were significant at P < .05. STATA software was used for analysis (release 13.0; StataCorp 2001, College Station, TX).
Results
Data source and general description
From 2002 to 2019, a total of 644 diagnoses of neutropenia were collected within the INR. The complete dataset of 214 patients with AN was retrieved from the registry, and 3 categories were defined according to the abovementioned criteria: pAN comprised 135 patients (63%), and LO-Np and LL-Np comprised 31 (15%) and 48 (22%) subjects, respectively.
Comparison of pAN vs LO-Np vs LL-Np
We initially compared these 3 groups to see whether there were differences besides age of onset and duration of neutropenia. Indeed the 3 populations showed significant differences in most of the analyzed variables with the exception of monocytosis, CD8+ cell depletion, deficiency of ≥1 class of immunoglobulin, and severity of infections that were similarly distributed among the 3 populations. Details are reported in Table 1.
Comparison between pAN, LO-Np, and LL-Np
| pAN (n = 135 patients) | LO-Np (n =31 patients) | LL-Np (n = 48 patients) | P | |
|---|---|---|---|---|
| Female sex | 41/135 (30) | 16/31 (52) | 25/48 (52) | .001 |
| Age at diagnosis, median (IQR), y | 0.6 (0.3-1.3) | 11.5 (7.6-14.6) | 1.18 (0.6-2.2) | <.001 |
| Lenght of neutropenia, median (IQR), y | 1.03 (0.54-1.7) | 2.1 (1.4-4.4) | 4.5 (3.5-7.09) | <.001 |
| Resolution of neutropenia | 135/135 (100) | 4/31 (13) | 28/48(58) | <.001 |
| ANC at onset, median (IQR), ×109/L | 0.43 (0.23-0.716) | 0.649 (0.43-0.97) | 0.552 (0.35-0.79) | <.001 |
| Leukocyte count at onset, median (IQR), ×109/L | 6.125 (5.01-7.92) | 3.18 (2.67-3.71) | 5.03 (3.44-6.90) | <.001 |
| Lymphocyte count, median (IQR), ×109/L | 4.74 (3.50-5.88) | 1.68 (1.24-1.90) | 2.37 (1.92-3.40) | <.001 |
| Monocitosis at onset | 15/120 (12.5) | 5/26 (19) | 10/38 (26) | NS |
| Low CD3* | 7/72 (10) | 2/24 (8) | 11/39 (28) | .02 |
| Low CD4* | 8/72 (11) | 4/25 (16) | 11/38 (29) | .06 |
| Low CD8* | 11/71 (15) | 4/25 (16) | 7/40 (17,5) | NS |
| Low CD19* | 6/64 (9) | 12/25 (48) | 13/37 (35) | <.001 |
| Low NK* (CD3+CD16+CD56+) | 10/62 (16) | 9/23 (39) | 10/34 (29) | .06 |
| Immunoglobulin depletion* | 7/113 (6) | 4/26 (15) | 3/44 (7) | NS |
| Infectious episodes | 65/130 (50) | 14/29 (48) | 18/47 (38) | .4 |
| Severe infections | 16/65 (25) | 3/14(21) | 3/18 (17) | .2 |
| G-CSF therapy | 7/135 (5) | 3/21 (14) | 7/42 (17) | .04 |
| Autoimmune diseases/markers | 2/135 (1) | 16/29 (55) | 8/48 (17) | <.001 |
| pAN (n = 135 patients) | LO-Np (n =31 patients) | LL-Np (n = 48 patients) | P | |
|---|---|---|---|---|
| Female sex | 41/135 (30) | 16/31 (52) | 25/48 (52) | .001 |
| Age at diagnosis, median (IQR), y | 0.6 (0.3-1.3) | 11.5 (7.6-14.6) | 1.18 (0.6-2.2) | <.001 |
| Lenght of neutropenia, median (IQR), y | 1.03 (0.54-1.7) | 2.1 (1.4-4.4) | 4.5 (3.5-7.09) | <.001 |
| Resolution of neutropenia | 135/135 (100) | 4/31 (13) | 28/48(58) | <.001 |
| ANC at onset, median (IQR), ×109/L | 0.43 (0.23-0.716) | 0.649 (0.43-0.97) | 0.552 (0.35-0.79) | <.001 |
| Leukocyte count at onset, median (IQR), ×109/L | 6.125 (5.01-7.92) | 3.18 (2.67-3.71) | 5.03 (3.44-6.90) | <.001 |
| Lymphocyte count, median (IQR), ×109/L | 4.74 (3.50-5.88) | 1.68 (1.24-1.90) | 2.37 (1.92-3.40) | <.001 |
| Monocitosis at onset | 15/120 (12.5) | 5/26 (19) | 10/38 (26) | NS |
| Low CD3* | 7/72 (10) | 2/24 (8) | 11/39 (28) | .02 |
| Low CD4* | 8/72 (11) | 4/25 (16) | 11/38 (29) | .06 |
| Low CD8* | 11/71 (15) | 4/25 (16) | 7/40 (17,5) | NS |
| Low CD19* | 6/64 (9) | 12/25 (48) | 13/37 (35) | <.001 |
| Low NK* (CD3+CD16+CD56+) | 10/62 (16) | 9/23 (39) | 10/34 (29) | .06 |
| Immunoglobulin depletion* | 7/113 (6) | 4/26 (15) | 3/44 (7) | NS |
| Infectious episodes | 65/130 (50) | 14/29 (48) | 18/47 (38) | .4 |
| Severe infections | 16/65 (25) | 3/14(21) | 3/18 (17) | .2 |
| G-CSF therapy | 7/135 (5) | 3/21 (14) | 7/42 (17) | .04 |
| Autoimmune diseases/markers | 2/135 (1) | 16/29 (55) | 8/48 (17) | <.001 |
Data are n/N (%) unless other noted. NS, not significant.
Values below the third percentile for age.
Median ANC values were significantly higher in LO-Np (0.649 × 109/L; interquartile range [IQR], 0.43-0.97) and in LL-Np (0.552 × 109/L; IQR, 0.376-0.79) vs pAN (0.43 × 109/L; IQR, 0.22-0.716) (Figure 1) and severe neutropenia (0.5 × 109/L) was more frequent in pAN (58%) vs LL-Np (37%) and LO-Np (32%), respectively (P = .03)
Lymphocytes were lower in LO-Np (1.68 × 109/L; IQR, 1.24-1.90) and LL-Np (2.37 × 109/L; IQR, 1.92-3.40) vs pAN (4.74 × 109/L; IQR, 3.50-5.88; P < .001). This might account for the higher frequency of leukopenia found in LO-Np (73%) vs LL-Np (26%) vs pAN (11%; P = .01).
Lymphocyte subset analysis showed that decreased CD19+ cells were more frequent in LO-Np (48%), followed by LL-Np (35%) and pAN (9%; P < .001) and so was depletion of CD3−CD16+CD56+ (natural killer [NK] cells; 39% in LO-Np), followed by LL-Np (29%) and by pAN (16%), although not definitely reaching statistical significance (P = .06).
The 3 populations also differed for the proportion of patients with low (absolute values; below the third percentile for age) CD3+ cells (P = .02). This difference was borderline for CD4+ cells (P = .06).
Rate of severe infections (ie, meningitis, sepsis, osteomyelitis, phlegmons or deep abscesses, pneumonia), as scored before G-CSF treatment, were equally distributed among the 3 groups and therefore were recurrent infections. Likewise, type of infections (fever of unknown origin and upper respiratory infections including otitis, skin infections) were comparable.
Despite a similar infectious load, G-CSF treatment, mostly on demand according to Italian National Guidelines,10 was adopted more frequently in LO-Np (3/21; 14%) and LL-Np (7/42; 17%) than in pAN (7/135; 5%; P = .04).
Finally, the occurrence during follow-up of autoimmune diseases was another element of differentiation among the 3 populations (Table 1). Both autoimmune markers and diseases occurred more frequently in LO-Np (55%) and LL-Np (17%) compared with pAN (1%; P = .01)
In particular, autoimmune diseases included the following, autoimmune thyroiditis (5 patients), Evans’ syndrome (4 patients), autoimmune lymphoproliferative syndrome (2 patients), type 1 diabetes (2 patients), celiac disease (1 patient), vitiligo (1 patient), rheumatoid arthritis (1 patient), and appeared 42 months (range, 5-168 months) since diagnosis of neutropenia, whereas autoimmune markers included anti–smooth muscle antibodies (3 patients), anti–double-stranded DNA (2 patients), antibodies to glutamic acid decarboxylase (1 patient), antithyroid antibodies (1 patient), and anti–Saccharomyces cerevisiae antibodies (1 patient).
Overall, this comparison indicates that, in our cohort, on immunohematologic and clinical ground, patients with pAN, LO-Np, and LL-Np differ not only because of late-onset or longer duration but also because of rate of resolution, leukopenia, severity of neutropenia, lymphopenia with low B+ and NK cells, increased need of G-CSF, and increased frequency of associated autoimmunity.
Comparison between LO and LL neutropenia
Because in the triple comparison of pAN vs LO-Np vs LL-Np, differences occurred also between the LO and LL groups, we next performed a paired analysis of these 2 populations for the same items of the triple evaluation (Table 1).
Whereas no significant differences were found in sex, monocytosis, severity of neutropenia, overall severe infection rate, need for G-CSF, and lymphocyte subsets, LO-Np in comparison with LL-Np appeared to have a lower rate of resolution (13% vs 58%; P < .001), lower white blood cells (WBC; 3.18 × 109/L vs 5.03 × 109/L; P < .001) and lymphocytes values (1.68 × 109/L in LO-Np vs 2.37 × 109/L in LL-Np; P < .001), higher occurrence of aphthae (24%; P = .008), and a stronger association with autoimmune diseases/markers (55% vs 17%; P = .001).
Although lymphocyte subset was nonsignificantly different in LO-Np vs LL-Np, a trend toward diminution of CD19+ and NK cells was seen in LO-Np compared with LL-Np, counterbalanced by CD3+ and CD4+ cells whose trend was opposite.
We applied an enlarged NGS panel including 315 genes involved in hematologic disorders, immunodeficiencies, immune dysregulation, inflammatory, and bone marrow failure syndromes to a small subgroup of LO-Np and LL-Np patients who showed some immune-related stigmata that are detailed in supplemental Table 1. We found variants in 4 of 15 tested patients (27%). Variants were detected in 3 of 10 LO-Np and in 1 of 5 LL-Np patients and were identified in TACI (2 in LO-Np, 1 variant of uncertain significance, and 1 likely pathogenic variant), TINF2 (1 variant of uncertain significance in LO-Np), and LRBA (2 compound heterozygosis of 2 pathogenic variants in LL Np) genes according to GnomAD and VarSome databases.
These findings raise the point that a clinical/biological difference might potentially exist between LO-Np and LL-Np, with LO-Np showing a more pronounced autoimmune signature.
Overall, considering the results of the triple (pAn vs LL-Np vs LO-Np) and of the double (LL-Np vs LO-Np) comparison, the issue that we are dealing with potentially different entities appears to be substantiated.
Discussion
The finding of autoimmune neutropenia patients apparently similar to classical pAN but indeed different from them because isolated neutropenia appears later than the age of 3 years (LO-Np) or because neutropenia persists beyond 36 months since diagnosis (LL-Np) is a rather common situation in clinical practice and represents a challenge for diagnosis and management because patients may not receive appropriate and tailored treatment and monitoring plans.
In our study, we describe a cohort of 79 autoimmune neutropenia patients who differ from classical pAN because either it presented after age of 3 years or recovered later than 36 months, and we compared this cohort with a group of pAN subjects whose diagnosis was firmly established, aiming to see whether they showed additional immunohematologic and clinical differences vs pAN. Indeed, this triple comparison showed that, in addition to late onset and longer duration, the 3 groups also differ in the rate of resolution, severity of neutropenia (that were both lowest in LO-Np), lymphopenia including reduced B+-cells (lowest in LO-Np) and low NK cells (although not fully statistically significant; P = .06), frequency of associated autoimmunity (whose highest rate was seen in LO-Np), and use of G-CSF compared with pAN. For reduced resolution rate, the difference was striking (100% in pAN vs 58% in LL-Np vs 13% in LO-Np), thus raising the hypothesis that a proportion of LL-Np patients still have a tendency to resolution that may occur later in time. It may be possible that these LL-Np patients are true pAN patients simply requiring a longer time beyond the classical 36 months to resolve their neutropenia. On the contrary, a small portion of subjects affected with those neutropenias are found mutated with monogenic immunodeficiency (TACI) or with disease including immune deficiency (TINF2).
Indeed, the differences we observed in the triple comparison were to a large extent seen also when we compared LO-Np with LL-Np, thus further corroborating the fact that we are probably dealing with different clinical entities. In fact, we found a significantly lower rate of resolution, total lower WBCs, and lymphocytes, and an increased rate of autoimmunity developing during the course of disease (median appearance time, 42 months since diagnosis of neutropenia) in LO-Np vs LL-Np. These findings indicate a more pronounced autoimmune signature for LO-Np compared with LL-Np, thus supporting the hypothesis that in LO-Np, neutropenia would be an anticipatory phenomenon of an immunodeficiency/dysregulation disease rather than an isolated event.4,5 Indeed primary immunodeficiencies are often associated to autoimmune phenomena, and it is also known that the immunologic defect and the exhaustion of immunoglobulin may occur in the third/fourth decade of life, which may explain the substantial lack of immunoglobulin deficiency we observed in our younger cohort. LL-Np and LO-Np that do not show resolution generally resemble the characteristics of AN of young adults. A possible hypothesis would be that neutropenia, being rather asymptomatic at the beginning, occurs as a precocious sign and is followed by acquisition of associated autoimmune signs over time. Prevalence of females in patients without resolution has been shown (P < .002), as generally described in AN of young adults.
LL-Np may appear to parallel what sometimes happens with immune thrombocytopenia,11 where those cases with long history or with poor response to first/second line immune thrombocytopenic purpura–directed therapy may anticipate lymphoproliferative (like chronic lymphocytic leukemia and large granular lymphocytic leukemia) or autoimmune disorders (ie, systemic lupus erythematosus). Interestingly, in the small aggregate group of LO-Np and LL-Np who underwent NGS analysis, we found variants in genes associated with monogenic immunodeficiency (TACI that encodes protein of the tumor necrosis factor receptor superfamily found on the surface of B cells, and LRBA, whose mutations are associated with common variable immunodeficiencies, autoimmunity, and susceptibility to inflammatory bowel disease) or with telomere biology diseases that also include immune deficiency (TINF2, encoding a component of the shelterin complex involved in telomere maintenance).12-15
This might be in keeping with the hypothesis that LO-Np may herald an immune deficiency or an immune dysregulation that will develop later over the disease time course.6 This may have clinical implications because patients, still resembling but not fully fitting classical diagnostic criteria for pAN, and who have later onset, lower WBC and lymphocyte count, no tendency to spontaneous resolution after 3 years since diagnosis, and have, although subtle, autoimmune stigmata, deserve long-term monitoring and extensive immunologic workup, including a large immune dysregulation NGS gene panel aimed to identify a later shift toward secondary AN or to an immune deficiency. This might apply also to that proportion of LL-Np who do not get disease resolution. Another relevant implication is that an early diagnosis of an immunologic disease may enable to prevent complication and endeavor the use of targeted therapies.16
We observed a more frequent G-CSF use in LO-Np and LL-Np vs pAN, although there was no difference of overall infectious rate between the 3 groups. This may be explained by the prevalent use of on-demand G-CSF in adherence to national guidelines10 that suggest temporarily use in case of otherwise unmanageable infections or as prophylaxis before surgery.
This study has some limitations. As a retrospective registry analysis, it implies some selection bias and some incomplete data as in the case of genetic tests. In addition, the relatively short follow-up does not allow to capture those patients who present in childhood with isolated neutropenia and that might become immunodeficient in adult age. In this respect, a joint study with adult neutropenia registry/groups would be advisable to fill this age gap.
There are, however, some important strengths represented by the robust comparative basis and the extensive clinical and immunohematologic characterization of the study populations.
Overall, this is the first large comparative study showing that children with apparent pAN but whose neutropenia lasts longer or appears later in life, have also other important differences, suggesting that cases presenting after 3 years of age, with low/no tendency to resolution, require tight monitoring and extensive immune investigations aimed to early identify underlying immunologic disease that might develop.
For data, please contact the corresponding author at francescafioredda@gaslini.org.
Acknowledgment
This work was supported by the Italian Ministry of Health (Ricerca Corrente 2020).
Authorship
Contribution: F.F. and G.A.R. designed the research; F.F., G.A.R., F.D., and P.F. performed research; A.T., A.B., E.M., L.L., L.N., M.P., F.V., G.R., S.B., G.B., A.F., M.V., U.R., D.O., B.M., R.G., S.L.D., and N.M. contributed to data collection; M.L., P.T., T.L., L.P., A.G., I.C., and F.B. performed laboratory analysis; S.Z., F.F., G.A.R., M.M., and C.D. reviewed and analyzed data; and F.F., G.A.R., and C.D. wrote the paper
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Francesca Fioredda, Unit of Hematology, IRCCS–Istituto Giannina Gaslini, Largo G Gaslini 5, 16147 Genoa, Italy; e-mail: francescafioredda@gaslini.org.

