

Key Points
Platelets harbor HIV in infected people prior to the initiation of antiretroviral therapy; thereafter, HIV+ platelets are reduced.
Infection of CD4+ T cells via HIV+ platelets is enhanced by platelet activation and subsequent platelet–T-cell complex formation.

Abstract
Platelets were recently found to harbor infectious HIV virions in infected individuals who are on antiretroviral treatment with poor CD4+ T-cell recovery. In this study, we screened platelets from recently infected individuals, before and after antiretroviral therapy, for the presence of virus and examined platelet activation, as well as CD4+ T-cell recovery. This was followed by in vitro studies assessing platelet–CD4+ T-cell complex formation as a contributing factor to viral transmission. HIV+ platelets were detected in 10 of 10 acutely infected individuals with no prior history of antiretroviral therapy. The percentage of HIV+ platelets dropped significantly after 3 months of antiretroviral therapy in all of the study participants. These individuals also demonstrated significant recovery of CD4+ T cells. Interestingly, the percentage of HIV+ platelets correlated positively with viral load but not with CD4+ T-cell count. Furthermore, we found that platelet activation with soluble CD40L or thrombin receptor activator peptide 6 (TRAP6) increased platelet-virus interactions in vitro. TRAP6-mediated interactions were reduced by platelet antagonists, aspirin, and R406. We demonstrated that platelets transmit the virus to CD4+ T cells, and this transinfection was abolished by inhibiting platelet–T-cell complex formation via exposure to an anti-CD62P antibody. Additionally, treatment with TRAP6 significantly increased the transinfection, which was also inhibited by aspirin and R206. These results reveal that platelets have the potential to promote HIV viral spread during the acute stage of infection, by harboring infectious virus transmitting infection to susceptible CD4+ T cells through complex formation.
Introduction
HIV-1 is the causative agent of AIDS, a disease that is characterized by the destruction of CD4+ T cells and the subsequent loss of adaptive immunity, leading to opportunistic infections. In 2018, it was estimated that 37.9 million people worldwide were living with HIV, with 1.7 million new diagnoses reported that year.1 It is estimated that 32 million people have died of AIDS-related illnesses since the beginning of the epidemic. However, because of the advent of combined antiretroviral therapy (cART), yearly AIDS-related deaths have decreased from 1.9 million, at their peak in 2005, to 770 000 in 2018.1 cART can also be used for pre- and postexposure prophylaxis to prevent acquisition of HIV.2-4
Although cART is effective in repressing viral loads and restoring the immune system, giving HIV-infected individuals a life expectancy close to normal,5 it is not perfect. Even in highly cART-adherent individuals, low levels of viremia can still be detected, signifying ongoing residual viral replication.6-10 People living with HIV also have persistent low-grade inflammation and immune activation11 and are at increased risk for developing diseases associated with inflammation, such as cardiovascular disease, renal failure, diabetes, and hypertension, as well as for developing these diseases at younger ages than the general population.12,13 Additionally, increased platelet activation has been well documented and is considered a hallmark of chronic HIV infection.14-17
Platelets have been shown to directly interact with HIV-1–infected cells in vivo. Platelets can form complexes with inflammatory monocytes and CD4+ T cells, and these complexes are found with increased frequency in HIV+ individuals.14,18 These interactions occur through binding of the P-selectin glycoprotein ligand-1 (PSGL-1) on CD4+ T cells or monocytes to P-selectin (CD62P) on platelets.14,18,19 CD62P is contained within platelet α granules and is only expressed on the cell surface following platelet activation.
Platelets have also been shown to interact directly with HIV-1 virions in vitro. Utilizing electron microscopy, platelets were shown to harbor HIV in their open canalicular system (OCS) and even to disseminate infection to susceptible cell lines.20,21 Similarly, a recent study by Banerjee et al reported that platelets engulfed fluorescent HIV pseudovirions and processed these virions inside of endosomal compartments.22 Cells of myeloid origin are also known to harbor HIV in plasma-membrane compartments that are analogous to the OCS in platelets.23-25 The believed mechanism behind this is the use of DC-SIGN and CLEC-2, 2 C-type lectins that are among a number of C-type lectins responsible for the same phenomenon in dendritic cells.26-28
However, few studies have examined the interaction of HIV with platelets in vivo. One study screened blood components for the presence of HIV, whereas another showed that platelet–red blood cell complexes could transfer infection to peripheral blood mononuclear cells. However, neither of these studies quantified this interaction or explained the mechanism whereby platelets may spread infection.29,30 A recent study suggests that platelets harbor infectious HIV particles and transmit virus to macrophages in infected people on cART who demonstrate poor CD4+ T-cell recovery.31 In this study, we sought to further examine whether platelets harbor infectious HIV virions during the acute phase of infection, to determine whether platelet-virus associations remain after efficacious cART and subsequent immune cell rebound, and to identify the role of platelet activation in this interaction. We hypothesized that platelets may act as an acute viral reservoir, prior to cART treatment, and aid in viral dissemination. Virus internalization by platelets may also serve as a form of immune evasion, as well as a way to augment infectivity, because platelets form complexes with virus-susceptible CD4+ T cells and monocytes. Thus, we additionally sought to identify whether complex formation facilitated viral infection.
Materials and methods
Ethics statement
The Research Subjects Review Board at the University of Rochester Medical Center approved studies involving human samples. All study participants were adults, and blood samples were obtained after written informed consent, in accordance with the Declaration of Helsinki.
Patient samples and platelet isolation
Blood samples from HIV− people (who had not taken anti-inflammatory medications within the prior 21 days) were collected into acid citrate dextrose–buffered Vacutainer tubes (BD Biosciences). Information on platelet isolation can be found in supplemental Materials and methods. All blood was processed within 2 hours of being collected.
Cryopreserved platelet-rich plasma samples were collected from people with HIV at 0 weeks (pre-cART) and at 3 months following initiation of cART, and 4 cryopreserved plasma samples were collected from HIV− individuals. Two additional cryopreserved plasma samples were collected only at the pre-cART time point. Additional cryopreservation methodology can be found in the supplemental Materials and methods. Patient demographics for HIV-infected individuals are outlined in Table 1. Viral loads were quantified from platelet-poor plasma following centrifugation of whole blood at 1500g for 5 minutes.
Demographics and clinical characteristics of study participants pre-cART and 3 months post-cART
| Characteristics | Data |
|---|---|
| Age, mean ± SD, y | 31.75 ± 8.2 |
| Race, n (%) | |
| African American/Black | 7 (58.3) |
| White | 4 (33.3) |
| Asian | 1 (8.4) |
| Sex, n (%) | |
| Male | 11 (91.6) |
| Female | 1 (8.4) |
| CD4 count, mean ± SD, cells/µL | |
| Pre-cART (n = 12) | 469 ± 234.3 |
| Post-cART (n = 10) | 510 ± 238 |
| Platelet count, mean ± SD, ×103cells/μL | |
| Pre-cART (n = 10) | 212.9 ± 65.8 |
| Post-cART (n = 10) | 221.1 ± 45.9 |
| Viral load | |
| Pre-cART (n = 12) | |
| HIV RNA log10 copies/mL, mean (range) | 3.6 (1.7-5) |
| Not reported, n (%) | 2 (16.6) |
| Post-cART (n = 10) | |
| HIV RNA log10 copies/mL, mean (range) | 2 (1.2-4.2) |
| Undetectable, n (%) | 4 (40) |
| Characteristics | Data |
|---|---|
| Age, mean ± SD, y | 31.75 ± 8.2 |
| Race, n (%) | |
| African American/Black | 7 (58.3) |
| White | 4 (33.3) |
| Asian | 1 (8.4) |
| Sex, n (%) | |
| Male | 11 (91.6) |
| Female | 1 (8.4) |
| CD4 count, mean ± SD, cells/µL | |
| Pre-cART (n = 12) | 469 ± 234.3 |
| Post-cART (n = 10) | 510 ± 238 |
| Platelet count, mean ± SD, ×103cells/μL | |
| Pre-cART (n = 10) | 212.9 ± 65.8 |
| Post-cART (n = 10) | 221.1 ± 45.9 |
| Viral load | |
| Pre-cART (n = 12) | |
| HIV RNA log10 copies/mL, mean (range) | 3.6 (1.7-5) |
| Not reported, n (%) | 2 (16.6) |
| Post-cART (n = 10) | |
| HIV RNA log10 copies/mL, mean (range) | 2 (1.2-4.2) |
| Undetectable, n (%) | 4 (40) |
Individuals with HIV (n = 12) were enrolled in this study, prior to any antiretroviral therapy. Individuals (n = 10) returned following 3 months of antiretroviral treatment. No individual in this study presented with opportunistic infections or other AIDS-defining illnesses at the time of enrollment.
Cells, viruses, and reagents
CD4+ T cells were isolated with CD4 MicroBeads (Miltenyi Biotec), according to the manufacturer’s instructions, and differentiated using IL-2 and CD3/28 Dynabeads (Gibco). More information can be found in supplemental Materials and methods.
HIV-1 BaL and IIIB were obtained from ZeptoMetrix.
In situ hybridization
Platelets from HIV+ cART-naive individuals were isolated as described above and fixed in 4% paraformaldehyde, washed with phosphate-buffered saline (PBS), and resuspended in 70% EtOH. Two hundred microliters of the suspension was mounted onto Superfrost Plus Microscope Slides (Fisher Scientific), through centrifugation at 1000 rpm for 10 minutes (Shandon Cytospin 4 Cytocentrifuge). In situ hybridization was performed using an RNAscope Multiplex Fluorescent v2 kit (ACD Bio) and HIV gagpol-C3 Probe (cat. no. 317691), in conjunction with Opal 570 dye (Akoya Biosciences), following the manufacturer’s instructions. Cells were counterstained with an anti-human CD61 FITC Antibody (BioLegend). Glass coverslips (Fisher Scientific) were mounted onto slides with ProLong Gold Antifade Mountant (Invitrogen), and slides were imaged at 100× with an Olympus FV1000 confocal microscope. Platelets from an HIV− donor were used as a negative control.
ImageStream analysis
Fixed platelets from HIV+ persons were permeabilized using Permeabilization Buffer (Invitrogen), blocked for 25 minutes in 2% bovine serum albumin, and probed using 1.25 ng/mL biotin-conjugated goat anti-gp120 (ab53937; Abcam) for 30 minutes. Platelets were stained using Streptavidin, Alexa Fluor 488 Conjugate (Invitrogen) and Alexa Fluor 647 anti-human CD61 Antibody (BioLegend) for 30 and 25 minutes, respectively, with 2 wash steps before and after each staining. Samples were acquired using an Amnis ImageStreamX imaging cytometer and analyzed (IDEAS, Amnis Corporation). The instrument and INSPIRE software were set up as follows: channels 01 (bright field), 02 (Alexa Fluor 488), 06 (bright field 2), and 11 (Alexa Fluor 647). Magnification was 60×, and lasers 488 and 658 were activated for fluorescence. The flow rate was set for low speed/high sensitivity; 105 events were recorded for each sample. Data analysis was performed by gating in-focus events using a frequency chart for Gradient RMS_M01_Ch01, then creating a scatter plot of aspect ratio and area to gate for platelet size, followed by gating for CD61+ cells. Glycoprotein 120 (gp120)+ cells were identified using the channel 02 Bright Detail Intensity setting. Cryopreserved platelets from HIV− donors were used as negative controls.
Super resolution microscopy
Platelets from cryopreserved plasma were isolated and probed for gp120 and CD61, as described above with some modifications. A Streptavidin 594 Conjugate (Invitrogen) was used in place of an Alexa Fluor 488 conjugate. Additionally, cells were stained with anti-mouse STAR RED (Abberior) and then subjected to 2 additional wash steps. Platelets were then plated on a poly-l-lysine (Millipore Sigma)–coated glass coverslip (Fisher Scientific), washed twice with PBS, mounted onto a glass slide (Fisher Scientific) with ProLong Gold Antifade Mountant (Invitrogen), and imaged at 100× (easy3D STED Microscope; Abberior). Cryopreserved platelets from an HIV− donor were used as a negative control.
Platelet-virus incubations and coculture
Platelets from HIV+ or HIV− people were isolated as described above. A total of 106 platelets isolated from HIV− donors was incubated with 2.25 ng (as defined by p24 levels) of HIV-1 BaL or HIV-1 IIIB for 1 hour and then washed 3 times. Tyrode’s Salt Solution was used for all wash steps.
For platelet-activation studies, platelets were incubated with 5 μM adenosine diphosphate (ADP) (Bio/Data Corporation), 1 μg/mL recombinant human soluble CD40L (sCD40L) (R&D Biosystems), 0.1 U/mL Thrombin from human plasma (Millipore Sigma), 10 μM thrombin receptor activator peptide 6 (TRAP6) (Tocris Bio-Techne), 1 μL dimethyl sulfoxide (DMSO) (Fisher Scientific), or control PBS for 30 minutes prior to the addition of virus. Additionally, platelets were incubated with a mouse immunoglobulin G2B (IgG2B) DC-SIGN–specific monoclonal antibody (cat. no MAB 1611) or a mouse IgG2B control (both from R&D Systems), at a final concentration of 10 μg/mL, and washed prior to the addition of virus. For platelet-inhibitor studies, platelets were incubated with 10 μg/mL aspirin, 0.5 μM R406, 10 μΜ ticagrelor, or DMSO for 20 minutes prior to the addition of TRAP6.
For platelet–T-cell cocultures, platelets were treated under the conditions described above or with 5 μg/mL CD62P neutralizing antibody 9E1 or mouse IgG1 control antibody (both from R&D Systems), as described.18 Platelets were washed and then plated with 7.5 × 105 CD4+ T cells at a 10:1 ratio. Negative controls included cART (10 μΜ each raltegravir, efavirenz, and tenofovir; National Institutes of Health AIDS Reagent Program) or adding the supernatant from the final platelet wash to cocultures. Twenty-four hours later, a 50-μL aliquot of suspension was removed and fixed in 2% paraformaldehyde, and platelet–T-cell complexes were measured by flow cytometry. At 72 hours, the media were removed and centrifuged at 1100g for 10 minutes and transferred to a new tube, twice. Virus was measured using a p24 Quantikine ELISA (R&D Systems), following the manufacturer’s instructions, and a SpectraMax iD3 plate reader (Molecular Devices).
Reverse transcriptase-polymerase chain reaction
Information on reverse transcriptase-polymerase chain reaction (RT-PCR) and quantitative RT-PCR can be found in supplemental Materials and methods.
Statistical analysis
GraphPad Prism 7 (GraphPad Software) was used for statistical analyses. Paired Student t tests were used to compare data for the percentages of gp120+ platelets pre- and post-cART. A 1-way analysis of variance (ANOVA) was used to analyze platelet-activation studies, and ratio Student t tests were used to compare treatment vs nontreatment conditions for p24 and CD62P mean fluorescence intensity of CD4+ cells cocultured with platelets and IgG vs CD4+ T cells with platelets and anti-CD62P antibody. Pearson or Spearman correlation tests were used to infer correlations, based on whether the data sets were parametric according to a D’Agostino-Pearson test and a Shapiro-Wilk normality test. All images were analyzed using Fiji (ImageJ; version 2.0.0).
Results
Antiretroviral therapy reduces viral uptake by platelets in persons with HIV
Previous in vitro coculture studies have identified the presence of HIV-1 or pseudotyped lentivirus in platelets, sequestered within the OCS or endosomal compartments, respectively.20-22,26,29,31,32 To verify whether platelets harbor virus in infected people, we isolated platelets from the plasma of viremic cART-naive donors and performed RNAscope probing for HIV gag genomic RNA, with subsequent immunostaining for CD61 to detect platelets. As shown in Figure 1A, platelets were found to contain viral genomic RNA. We further confirmed viral uptake by platelets through the use of super resolution microscopy, immunostaining for the HIV envelope glycoprotein, gp120, in concert with CD61. Here, we detected gp120-containing particles that were comparable in size to HIV virions (Figure 1B).33-35

Antiretroviral therapy reduces viral uptake by platelets in persons with HIV. (A) Platelets were isolated from the plasma of viremic cART-naive individuals, RNAscope probing was done for the HIV gag gene (green), followed by immunostaining for CD61 as a platelet marker (red). Images were taken using a confocal microscope (60× objective). Results are representative of 2 donors. Platelets isolated from an HIV− individual were used as a negative staining control (lower panels). (B) Isolated platelets were immunostained for viral glycoprotein gp120, in addition to CD61, and imaged using super resolution microscopy. (C) Platelets were imaged using ImageStream flow cytometry and gated using CD61 and gp120. (D) The percentage of gp120+ platelets was determined prior to cART and 3 months following cART in 10 matching samples. Significance between groups was calculated using a Wilcoxon matched-pairs signed-rank test. (E) Correlation between viral load and percentage of gp120+ platelets was examined in samples with detectable viral loads. Correlation was calculated by nonparametric Spearman correlation.
Antiretroviral therapy reduces viral uptake by platelets in persons with HIV. (A) Platelets were isolated from the plasma of viremic cART-naive individuals, RNAscope probing was done for the HIV gag gene (green), followed by immunostaining for CD61 as a platelet marker (red). Images were taken using a confocal microscope (60× objective). Results are representative of 2 donors. Platelets isolated from an HIV− individual were used as a negative staining control (lower panels). (B) Isolated platelets were immunostained for viral glycoprotein gp120, in addition to CD61, and imaged using super resolution microscopy. (C) Platelets were imaged using ImageStream flow cytometry and gated using CD61 and gp120. (D) The percentage of gp120+ platelets was determined prior to cART and 3 months following cART in 10 matching samples. Significance between groups was calculated using a Wilcoxon matched-pairs signed-rank test. (E) Correlation between viral load and percentage of gp120+ platelets was examined in samples with detectable viral loads. Correlation was calculated by nonparametric Spearman correlation.
Having shown that genomic HIV-1 RNA and HIV-1 envelope glycoprotein were associated with platelets from infected persons, we needed to quantify the percentage of platelets harboring HIV. We performed imaging cytometry on platelets immunostained for gp120 and CD61 (Figure 1C). As part of this analysis, we screened matching samples from HIV-infected individuals prior to and 3 months following the initiation of cART. As indicated in Table 1, after 3 months of cART, these subjects demonstrated significant suppression of viremia and corresponding recovery of CD4+ T cells. Prior to cART, 0.11% to 4.80% of platelets contained gp120; this percentage decreased significantly to 0.01% to 0.44% following cART initiation (Figure 1D). This was comparable to the numbers of HIV+ platelets reported previously in cART-treated HIV-infected subjects who presented poor CD4+ T-cell recovery.31 The percentage of gp120+ platelets from all HIV+ samples correlated with the individual viral load, although no correlation was observed when samples were divided by treatment status (Figure 1E; supplemental Figure 1A-B). Additionally, we did not observe a correlation between the percentage of gp120+ platelets and CD4+ T-cell count (supplemental Figure 1C). Within this cohort, only 4 of the 10 HIV+ donors had undetectable viral loads after 3 months of cART; however, there was not a significant difference in the percentage of gp120+ platelets prior to cART therapy in people who achieved an undetectable viral load compared with those with a detectable viral load after 3 months (Table 1; supplemental Figure 1D).
These data suggest that platelets naturally take up HIV virions in infected individuals and that, although cART decreases the viral uptake, virus-platelet complexes may remain present after 3 months of treatment.
Effect of platelet activators on HIV-platelet interaction
To confirm previous findings that platelets harbor HIV in vitro,20,21,26,31 we incubated platelets from HIV− individuals with 2.25 ng of HIV-1 BaL, an R5-tropic virus. Transmission electron microscopy showed virion-like structures within platelets (supplemental Figure 2A, right panel), presumably in their OCS or endosomes.20-22 Virion structure and size were confirmed by comparing these images with those of viral stocks (supplemental Figure 2A, left panel). Furthermore, we used RNAscope, probing for HIV gag genomic RNA and CD61 (Figure 2A). To test whether platelet-virus interactions are associated with selective coreceptor (CCR5 or CXCR4) usage, we incubated platelets with HIV-1 BaL (R5-using) or HIV-1 IIIB (X4-using) virus stocks and performed RT-PCR to detect the HIV tat gene in platelets. We identified genomic tat RNA in platelets exposed to both viruses (supplemental Figure 2B), suggesting that such interactions are not coreceptor restricted, which is consistent with previously reported studies.26
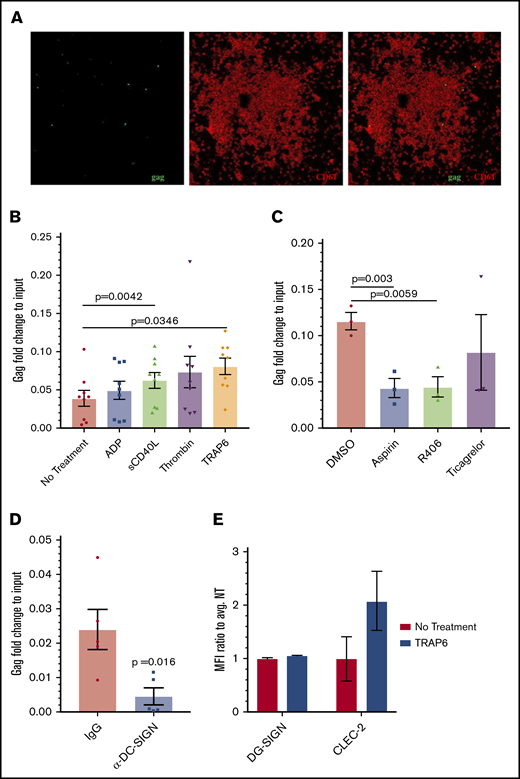
Effect of platelet activators on HIV-platelet interactions. (A) RNAscope probing for the HIV gag gene (green), followed by immunostaining for CD61 as a platelet marker (red) was also performed on these platelets. Images were taken using a confocal microscope (60× objective) and are representative of 2 independent trials. (B) Isolated platelets were treated with ADP (5 μM), sCD40L (1 μg/mL), thrombin (0.1 U/mL), or TRAP6 (10 μM) prior to the addition of HIV-1 BaL. Quantitative RT-PCR for HIV gag was performed and normalized to RNA extracted from the same viral stock. Data were analyzed by 1-way ANOVA. (C) Isolated platelets were treated with aspirin (10 μM), Syk inhibitor R406 (0.5 μM), ticagrelor (10 μΜ), or vehicle control prior to the addition of TRAP6 and HIV BaL-1; data were analyzed using 1-way ANOVA. (D) Platelets were treated with an IgG control or anti–DC-SIGN antibody prior to incubation with HIV-1 BaL. A ratio-paired Student t test was used to determine significance. (E) DC-SIGN and CLEC-2 expression was analyzed by flow cytometry. Mean fluorescence intensity (MFI) of DC-SIGN/CLEC-2 was measured on CD61-gated cells and normalized to the average MFI of the control.
Effect of platelet activators on HIV-platelet interactions. (A) RNAscope probing for the HIV gag gene (green), followed by immunostaining for CD61 as a platelet marker (red) was also performed on these platelets. Images were taken using a confocal microscope (60× objective) and are representative of 2 independent trials. (B) Isolated platelets were treated with ADP (5 μM), sCD40L (1 μg/mL), thrombin (0.1 U/mL), or TRAP6 (10 μM) prior to the addition of HIV-1 BaL. Quantitative RT-PCR for HIV gag was performed and normalized to RNA extracted from the same viral stock. Data were analyzed by 1-way ANOVA. (C) Isolated platelets were treated with aspirin (10 μM), Syk inhibitor R406 (0.5 μM), ticagrelor (10 μΜ), or vehicle control prior to the addition of TRAP6 and HIV BaL-1; data were analyzed using 1-way ANOVA. (D) Platelets were treated with an IgG control or anti–DC-SIGN antibody prior to incubation with HIV-1 BaL. A ratio-paired Student t test was used to determine significance. (E) DC-SIGN and CLEC-2 expression was analyzed by flow cytometry. Mean fluorescence intensity (MFI) of DC-SIGN/CLEC-2 was measured on CD61-gated cells and normalized to the average MFI of the control.
Next, we treated platelets with platelet agonists ADP (mild activator), sCD40L (mild activator), TRAP6 (intermediate activator), and thrombin (strong activator) prior to their incubation with HIV-1 BaL. We confirmed platelet activation using flow cytometry and staining for CD62P, a membrane-bound protein stored in platelet α granules that is expressed on the surface following platelet activation (supplemental Figure 2C). Under these conditions, platelet interactions with HIV were measured using quantitative RT-PCR to quantify genomic HIV gag RNA. Although all of the platelet agonists increased platelet-virus associations, only sCD40L and TRAP6 treatments reached statistical significance (Figure 2B). We did not observe a correlation between viral uptake and overall level of CD62P surface expression (supplemental Figure 2D), suggesting that this process might be secondary to the signaling pathways activated and subsequent changes in platelet infrastructure by these reagents,36-38 rather than the activation status of platelets per se. Treatment with platelet inhibitors aspirin and R406 prior to the addition of TRAP6 decreased the amount of associated virus, whereas pretreatment with ticagrelor did not (Figure 2C). None of these treatments significantly decreased surface expression of CD62P (supplemental Figure 2E). Interestingly, pretreatment with DMSO, which has been shown to inhibit platelet activation,39,40 decreased the amount of associated virus as well (supplemental Figure 2F). Anti–DC-SIGN antibodies were previously shown to decrease this interaction and were used here as a positive control (Figure 2D).26
Because DC-SIGN and CLEC-2 have been implicated for HIV attachment to platelets in previous studies,26,29 and we observed that pretreatment with anti–DC-SIGN antibodies ablated platelets binding HIV (Figure 2D), we tested whether treatment with TRAP6 increased surface expression of these C-type lectins. Interestingly, although we did not observe any change in DC-SIGN expression, there was an increase in surface-bound CLEC-2 on platelets following treatment with TRAP6 (Figure 2E).
Platelet-bound HIV infects CD4+ T cells through platelet–T-cell complex formation
Although previous studies have reported that platelet-associated HIV can infect CD4+ T cells under coculture conditions,26,29 it is unknown whether such infection occur directly through platelet–T-cell complex formation. To address this question, we isolated and differentiated CD4+ T cells from HIV− donors. Platelets isolated from cART-naive viremic donors were treated with an anti-CD62P antibody to prevent CD62P-PSGL-1–driven complex formation or an IgG1 control antibody. Treatment with the anti-CD62P antibody sharply reduced platelet–CD4+ T cell complex formation (Figure 3A). Additionally, this inhibition significantly decreased viral replication at 72 hours postcoculture, as measured by HIV-1 p24 levels in culture supernatants (Figure 3B). Because platelets lack a nucleus, it is highly unlikely that the de novo viral replication in this experiment occurred in platelets. This notion was further supported by the fact that p24 levels correlated significantly with platelet–T-cell complex formation (supplemental Figure 3A). Together, these data suggest that complex formation is required for efficient transfer of infectious virus to CD4+ T cells.

Platelet-bound HIV infects CD4+T cells through platelet–T-cell complex formation. CD4+ T cells were isolated from HIV− donors and differentiated using anti-CD3/CD28 beads and IL-2. Platelets from viremic cART-naive donors were treated with anti-CD62P or IgG control antibody and then cocultured with differentiated T cells. Flow cytometry was performed by staining for CD4 and CD61. (A) Platelet–T-cell complexes were then detected by flow cytometry using antibody specific for CD4 and CD61. Mean fluorescence intensity (MFI) of CD61 was used on CD4 gated cells as a measure of platelet–T-cell complex formation at 24 hours. Correlations were calculated using a Pearson correlation. (B) HIV-1 p24 was quantified by enzyme-linked immunosorbent assay (ELISA) in cell-free culture supernatants 72 hours after coculture (n = 3). Significance was determined using a paired Student t test. (C) Platelets from HIV− donors were incubated with HIV-1 IIIB and then treated with anti-CD62P or IgG antibodies, as above, prior to coculture with differentiated CD4+ T cells. CD4+ CD61 MFI was used to measure platelet–T-cell complex formation. (D) HIV-1 p24 was quantified by ELISA in cell-free supernatant at 72 hours postcoculture (n = 4). (E) Platelets from HIV− donors were treated with aspirin, R406, anti–DC-SIGN antibodies, or DMSO control prior to treatment with TRAP6 or vehicle control and subsequent incubation with HIV-1 IIIB prior to coculture with differentiated CD4+ T cells. Cocultures were treated with cART or supernatant from the third platelet wash as negative controls. HIV p24 was quantified by ELISA in cell-free supernatant at 72 hours after coculture (n = 4).
Platelet-bound HIV infects CD4+T cells through platelet–T-cell complex formation. CD4+ T cells were isolated from HIV− donors and differentiated using anti-CD3/CD28 beads and IL-2. Platelets from viremic cART-naive donors were treated with anti-CD62P or IgG control antibody and then cocultured with differentiated T cells. Flow cytometry was performed by staining for CD4 and CD61. (A) Platelet–T-cell complexes were then detected by flow cytometry using antibody specific for CD4 and CD61. Mean fluorescence intensity (MFI) of CD61 was used on CD4 gated cells as a measure of platelet–T-cell complex formation at 24 hours. Correlations were calculated using a Pearson correlation. (B) HIV-1 p24 was quantified by enzyme-linked immunosorbent assay (ELISA) in cell-free culture supernatants 72 hours after coculture (n = 3). Significance was determined using a paired Student t test. (C) Platelets from HIV− donors were incubated with HIV-1 IIIB and then treated with anti-CD62P or IgG antibodies, as above, prior to coculture with differentiated CD4+ T cells. CD4+ CD61 MFI was used to measure platelet–T-cell complex formation. (D) HIV-1 p24 was quantified by ELISA in cell-free supernatant at 72 hours postcoculture (n = 4). (E) Platelets from HIV− donors were treated with aspirin, R406, anti–DC-SIGN antibodies, or DMSO control prior to treatment with TRAP6 or vehicle control and subsequent incubation with HIV-1 IIIB prior to coculture with differentiated CD4+ T cells. Cocultures were treated with cART or supernatant from the third platelet wash as negative controls. HIV p24 was quantified by ELISA in cell-free supernatant at 72 hours after coculture (n = 4).
We repeated this experiment using platelets from HIV− donors that had been incubated with infectious virus in vitro. Treating platelets with the anti-CD62P antibody prior to coculture decreased platelet–CD4+ T-cell complex formation (Figure 3C) and the amount of p24 in culture supernatants (Figure 3D), although it did so to a lesser degree than observed with platelets from viremic individuals (Figure 3B). Nevertheless, supernatant p24 levels correlated with platelet–T-cell complex formation (supplemental Figure 3B). Collectively, these data confirm that platelets from infected individuals can spread infection to CD4+ T cells and that this process is greatly enhanced by platelet–T-cell complex formation. To understand the role of platelet activation, aside from its contribution to platelet–T-cell complex formation, we tested the contribution of platelet inhibitors and TRAP6 to the degree of transinfection. TRAP6 significantly increased transinfection of CD4+ T cells, and platelet inhibitors significantly hindered TRAP6-driven increases (Figure 3E). As reported previously,26,29 pretreating platelets with an anti–DC-SIGN antibody decreased transinfection, cART completely blocked viral replication, and no infection occurred from culturing CD4+ T cells with supernatants from the final platelet wash step, suggesting that infections were not caused by residual virus.
Discussion
Platelets’ ability to harbor infectious HIV-1 virions has been reported in vitro and in infected patients. This association may serve as a viral reservoir during acute stages of infection and facilitate viral dissemination through the blood and into immunologically privileged sites.41 However, driving factors, such as platelet activation status and the mechanism behind the transfer of virus to susceptible cells, have not been elucidated; also, the extent to which platelets associate with virus in the blood of infected persons, before and after cART, has not been quantified. Here, we show that HIV-associated platelets are found in cART-naive viremic individuals and are reduced, but not entirely ablated, following 3 months of cART. Additionally, platelets from cART-naive viremic patients can spread infection to differentiated CD4+ T cells directly through PSGL-1-CD62P–driven complex formation. TRAP6-induced platelet activation significantly increased transinfection, yet was drastically reduced by aspirin and R406 treatment. Together, these data signify that platelet activation may play a role in the dissemination of platelet-bound HIV through increased viral uptake, transinfection, and complex formation with susceptible cells. Importantly, platelet inhibitors can significantly reduce platelet-virus associations and the resulting contribution to viral spread, signifying a potential to improve treatment during the acute stage of initial infection.
HIV-associated platelets have been identified in infected persons through polymerase chain reaction, and platelet–red blood cell complexes have been shown to spread infection to cultured peripheral blood mononuclear cells.29,30 We report that a significant number of platelets from cART-naive viremic individuals were associated with HIV-1 genomic RNA and gp120 envelope glycoprotein. This association was correlated with viral load, and it decreased significantly following 3 months of cART; however, it was still present in persons with a detectable viral load. Interestingly, such a decrease in the percentage of HIV+ platelets did not have any correlation with the CD4+ T-cell count following cART; thus, it seems unlikely that CD4+ T cells play a role in the clearance of infected platelets.
Platelet infection could result from active thrombopoiesis of HIV-infected megakaryocytes, as recently suggested,31 or via spontaneous uptake of HIV virions by platelets in the circulation of viremic patients, because subsets of platelets express DC-SIGN and CLEC-2.20,26 It is possible that both events contribute to HIV+ platelets in viremic individuals, resulting in higher levels of platelet-bound virions pre-cART, and only a decrease, not eradication, following treatment. These reports and our data further suggest that platelets likely harbor infectious HIV particles in their OCS, internal membrane structures that make up a tunneling network of surface-connected channels, and would subsequently shelter the virus from the hostile immune environment. Although other in vitro studies utilizing pseudotyped lentiviruses identified virions inside platelet endosomes that were eventually degraded,22,32 our data, as well as other studies that used wild-type virus, show that platelet-associated virus is spread to other cells, indicating that the virus is not degraded.21,26,29,31 An additional in vitro study found that platelet-bound virus remained infectious for 2 to 3 days,26 which is a sufficient amount of time for platelets to facilitate viral spread as they travel throughout the circulatory system. This, in turn, suggests that platelets act as an acute reservoir, as well as associate with virus during the course of infection, as long as there is virus present in the blood or infected megakaryocytes. Additionally, ongoing cART does not prevent this association from occurring.
Interestingly, transmission of virus from 2 distinct sets of platelets, those from viremic patients or infected in vitro, to differentiated CD4+ T cells appears to depend differentially on CD62P-mediated complex formation (Figure 3D). This discrepancy may be because, unlike platelets derived from viremic patients, the naive platelets were exposed to virus in vitro for only 1 hour in this experiment, potentially resulting in weak/transient association of virions to the platelet and allowing for complex-independent transfer of virus from platelets to T cells. If a similar phenomenon were to occur in vivo, it is possible that platelets recently exposed to HIV could spread virus through complex formation and by releasing weakly bound virus. During acute infection, HIV has been detected in the cerebrospinal fluid of patients as early as 8 days following exposure.41 Platelets may aid in the rapid dissemination of virus throughout the body through shuttling infectious virions throughout the blood and passing on infection to susceptible cells.
We also found that platelet activation enhances viral uptake, as well as facilitates the transfer of infectious virus from platelets to susceptible CD4+ T cells, in part because activated platelets express surface-bound CD62P, which drives platelet–CD4+ T-cell complex formation. Apart from complex formation, platelets activated with TRAP6 had an enhanced ability to bind and transfer infectious virions. This is significant because HIV-infected individuals have increased levels of platelet activation,15-17 as well as increased platelet–CD4+ T-cell and platelet-monocyte complexes.14,18 Although treatment with platelet inhibitors aspirin and R406 decreased engulfment of virions and subsequent spread of infection, ticagrelor did not. Aspirin and R406 block COX-1 and Syk signaling, respectively, whereas ticagrelor inhibits P2Y12, the receptor for ADP, which we found to have little effect on platelet uptake of HIV. Only TRAP6 and sCD40L significantly increased viral uptake. Additionally, we did not observe an associated decrease in platelet–T-cell complex formation after 24 hours of treatment with platelet inhibitors. One explanation for this is that, although aspirin and R406 decreased platelet uptake of infectious virus, they did not drastically decrease CD62P expression. Collectively, these data imply that engulfment is increased following specific activation signals, as opposed to cellular activation in general. Studies have also reported that chemokines released in platelet α granules can enhance CD4+ T-cell infection.42,43 However, CXCL4/PF4 was previously noted to suppress viral infection, TRAP6-activated platelets may have exhausted inhibitory cytokines, whereas platelets pretreated with inhibitors may have had a delayed release of chemokines.44 PSGL-1-CD62P–driven complex formation is also responsible for the formation of platelet-monocyte complexes; it is possible that this may contribute to HIV dissemination to monocytes, thereby resulting in neuroinflammation.45-47 Therefore, further studies to determine whether platelet-bound virus can contribute to viral neuroinvasion would be valuable.
In summary, we have shown that platelets harbor HIV in infected individuals and that this interaction remains detectable, even after 3 months of cART. Platelet activation, particularly by sCD40L and TRAP6, further increases this virus-platelet association with TRAP6 treatment, also enhancing transinfection of CD4+ T cells. Finally, our data also demonstrate that platelet–CD4+ T-cell complex formation drives CD4+ T-cell infection by platelet-bound virus, suggesting that platelets may function as acute viral reservoirs and promote the spread of HIV in infected individuals.
Data sharing requests should be sent to Sanjay B. Maggirwar (smaggirwar@e-mail.gwu.edu).
Acknowledgments
The authors thank the Rochester Victory Alliance (Rochester, NY) for help in recruiting study subjects. They also thank Karen Bentley (University of Rochester Electron Microscopy Core), Kaye Thomas (University of Rochester Confocal and Conventional Microscopy Core), and Wojciech Wojciechowski (University of Rochester Flow Cytometry Core) for technical assistance.
This work was supported by grants from the National Institutes of Health (NIH) (R01 HL128155 [National Heart, Lung, and Blood Institute] and R01 NS066801 [National Institute of Neurological Disorders and Stroke]) (S.B.M.) and the University of Rochester Center for AIDS Research (P30 AI078498 from the NIH, National Institute of Allergy and Infectious Diseases [NIAID]). The clinical data and biospecimen were derived from NIH, National Institute of Mental Health (MH099921) support to G.S. S.R.S. was supported by HIV Pathogenesis and Replication Training Grant T32 AI049815 from the NIH, NIAID.
Authorship
Contribution: S.R.S. and S.B.M. conceived the project; S.R.S., M.V.S., and S.B.M. designed the study; S.R.S. performed experiments, analyzed results, created the figures, and wrote the manuscript; and S.D. and G.S. provided clinical data and biospecimens, advised the study, and assisted with writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sanjay B. Maggirwar, Department of Microbiology, Immunology, and Tropical Medicine, The George Washington University School of Medicine and Health Sciences, 2300 I St NW, 502A, Washington, DC 20052; e-mail: smaggirwar@email.gwu.edu.

