

Abstract
The majority of historical therapies for managing T-cell lymphomas (TCLs) have consisted of T-cell–depleting strategies. Unfortunately, these forms of therapies can hamper the ability to mount effective antitumor immune responses. Recently, the use of checkpoint inhibitors has revolutionized the therapy of solid and hematologic malignancies. The development of immunotherapies for the management of TCL has lagged behind other malignancies given 2 central reasons: (1) the competing balance of depleting malignant T cells while simultaneously enhancing an antitumor T-cell response and (2) concern for tumor hyperprogression by blocking inhibitory signals on the surface of the malignant T cell, thereby leading to further proliferation of the malignant cells. These challenges were highlighted with the discovery that programmed cell death protein 1 (PD-1) functions paradoxically as a haploinsufficient tumor suppressor in preclinical TCL models. In contrast, some preclinical and clinical evidence suggests that PD-1/programmed death ligand 1 may become an important therapeutic tool in the management of patients with TCL. Improved understanding of the immune landscape of TCL is necessary in order to identify subsets of patients most likely to benefit from checkpoint-inhibitor therapy. With increased preclinical research focus on the tumor microenvironment, substantial strides are being made in understanding how to harness the power of the immune system to treat TCLs. In this review, designed to be a “call to action,” we discuss the challenges and opportunities of using immune-modulating therapies, with a focus on checkpoint inhibitors, for the treatment of patients with TCL.
Introduction
Mature T-cell lymphomas (TCLs) are heterogenous diseases that include 27 different subtypes according to the 2016 World Health Organization (WHO) classification,1 making up ∼10% to 15% of non-Hodgkin lymphma (NHL) cases.2 TCLs can colloquially be categorized as peripheral TCLs (PTCLs) or cutaneous TCLs (CTCLs) and will be referenced accordingly throughout this review.1,3,4 The most common PTCLs are anaplastic large cell lymphoma (ALCL), follicular helper TCLs (inclusive of angioimmunoblastic TCL), and PTCL not otherwise specified (frequently referred to as PTCL-NOS). The most common CTCLs are mycosis fungoides (MF), Sézary syndrome (SS), and primary cutaneous CD30+ lymphoproliferative disorders.1
First-line therapy for PTCLs frequently consists of aggressive multiagent chemotherapy, often followed by autologous stem cell transplant consolidation.5 Analogous to treatment of aggressive B-cell lymphomas, iterations of a cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) backbone are commonly used in the first-line management of PTCLs. To improve upon these outcomes, more intensive combination chemotherapy approaches, such as CHOP plus etoposide (CHOEP) and dose-adjusted, continuous infusional dosing of etoposide, prednisone, vincristine, doxorubicin, and bolus dosing of cyclophosphamide (DA-EPOCH) have been used. Based on retrospective and post hoc analyses, outcomes appear improved in PTCL patients treated with etoposide-containing regimens,6-8 though prospective validation is needed. Recently, the ECHELON2 phase 3 trial, comparing CHOP vs brentuximab with cyclophosphamide, doxorubicin, prednisone (CHP) in CD30+ PTCL patients, demonstrated improved survival with brentuximab plus CHP. In ECHELON2, CD30+ was defined as >10% CD30 expression in the tumor biopsy by immunohistochemistry; the study enrolled 75% ALCL patients. CD30+ TCLs encompass ∼30% of all PTCLs; how much outcomes are improved with brentuximab plus CHP in CD30+ non-ALCL patients remains uncertain.9 As a result of ECHELON2, brentuximab plus CHP is considered category 1 first-line treatment of ALCL and a preferred regimen for other CD30+ PTCLs in the National Comprehensive Cancer Network (NCCN) guidelines.
In contrast to most PTCLs, early-stage CTCLs with low burden of disease in the skin typically have an indolent presentation and the disease can be controlled with skin-directed therapies for many years. A substantial fraction of patients will either progress on skin-directed therapy or develop more advanced-stage disease requiring systemic therapies. All consensus guidelines emphasize that CTCLs are chronic diseases with a relapsing course and the main goals of therapy are disease control, effective symptom management, and prompt treatment of life-threatening disease.10 Selection of therapy is typically a stage-based approach initially moving from least toxic skin-directed therapies to IV therapies with increasing toxicity.11 Efficacy of systemic therapies is suboptimal with response rates to most single-agent treatments below 50%.12 All therapies are considered palliative, and the only potentially curative therapy is an allogenic hematopoietic cell transplant.
Within the past decade, the development of novel immunotherapies has revolutionized the management of many solid and hematologic malignancies. By harnessing the immune system to treat cancer, unprecedented numbers of patients with advanced malignancies are experiencing durable remissions with substantially fewer side effects than what is seen with traditional cytotoxic chemotherapy. As a result of this success, there is unprecedented interest in understanding the role of immune dysfunction in cancer progression. One example of an immunotherapeutic approach that has shown efficacy as a single agent in a variety of cancers, including in patients heavily pretreated with TCLs, is checkpoint blockade (CPB).13 The programmed death (PD) pathway serves as a checkpoint to limit T-cell–mediated immune responses. Blocking the programmed cell death protein 1 (PD-1) receptor on the T cell results in T-cell activation and proliferation, inducing a potent immunotherapeutic antitumor effect.
A protumor microenvironment promotes development and progression of TCL
Within the tumor microenvironment (TME) of lymphomas exists a diverse repertoire of immune cells, including macrophages, B and T lymphocytes, and plasma cells.14,15 This immune diversity is best characterized in Hodgkin lymphomas, where the malignant Reed-Sternberg cell is relatively rare compared with the rich immune infiltrate. Similarly, the malignant T cell may not be the predominant immune cell type in a TCL tumor biopsy specimen.16,17 An immune-suppressive microenvironment is expected to play a significant role in the etiopathogenesis and progression of TCLs.18
Perhaps the best-characterized component of the TME in solid and hematologic malignancies is the tumor-infiltrating lymphocyte (TIL). The majority of TILs are CD8+ T cells, which engage with major histocompatibility complex class I (MHC I) on tumor cells via the T-cell receptor (TCR).19 In the presence of appropriate costimulation, for example, CD28 on the T cell interacting with B7-1 or B7-2 on the tumor cell or antigen-presenting cell (APC), a T-cell cytolytic response is triggered that is characterized by release of perforin and granzyme B. Inhibitory receptors, also known as checkpoints, the best characterized of which are CTLA-4 and PD-1, function to limit the proliferation and activity of cytolytic T cells. CTLA-4 is a surface protein expressed on CD4+ and CD8+ T cells (and to a greater extent on CD4+CD25+ T-regulatory cells20 ) that binds with a higher affinity to B7 molecules on APCs relative to CD28.21 When engaged to B7 molecules, CTLA-4 raises the threshold needed for T-cell activation and arrests T-cell differentiation.21
The TCR recognizes antigens presented by APCs, such as dendritic cells, which can lead to T-cell proliferation. In the majority of CTCLs, the malignant T cell is a clone of a CD4+ T cell.22,23 Dendritic cells are in rich abundance in CTCL lesions, and when malignant cells of patients with leukemic-phase SS are cocultured with dendritic cells, the TCR interaction of the malignant CD4+ T cell with MHC class II on dendritic cells sustains long-term proliferation of the tumor cells.22,24 The sustained engagement of TCR with antigens presented by dendritic cells is thought to contribute to an exhausted phenotype of T cells (both malignant and nonmalignant) within the TME, characterized by increased expression of PD-1, lymphocyte-activation gene 3 (LAG-3), T-cell immunoglobulin mucin-3 (TIM-3), and CTLA-4.25
Macrophages also have a relatively well-characterized role within the TME of TCLs. M1 macrophages tend to have a proinflammatory phenotype and are induced by T-helper 1 (Th1)-secreted cytokines such as interferon γ and tumor necrosis factor α (TNF α). M1 macrophages are the predominant form in early carcinogenesis. On the other hand, M2 macrophages generally promote an anti-inflammatory and proangiogenic phenotype. M2 macrophages are the predominant form in advanced carcinogenesis and are thought to contribute to an immune-suppressive TME.26 M2 macrophages comprise the predominant fraction of tumor-associated macrophages (TAMs). Similar to what is observed in various solid tumors, TCLs with increased M2 infiltration tend to be at an advanced stage and have a worse prognosis.27,28 Preclinically, it has been demonstrated that TAM depletion using clodronate‐encapsulated liposomes29 leads to delayed development of CTCL.30 Clodronate treatment reduced angiogenesis and lymphangiogenesis in these models, reaffirming the role of M2 macrophages in the regulation of vascular endothelial growth factor signaling.31 The clodronate-containing liposomes were not directly toxic to human Sézary cells in vitro, suggesting the antitumor effects were mediated by depletion of macrophages within the TME.30 Of clinical relevance, it has been shown that M2 CD163+ macrophages abundantly express CD30, a finding that suggests brentuximab’s antitumor activity may in part be secondary to architectural changes in the TME and may help explain reports of brentuximab vedotin activity in CD30− lymphoma patients.32
Chemotherapy may promote an antitumor immune response in TCL
Although chemotherapy was historically believed to be immune-suppressive, there is an increasing interest in understanding the role of the immune system in mediating chemotherapy’s antitumor efficacy. In response to chemotherapy, there are changes in the tumor-immune microenvironment; in favorably responding patients, an infiltration of T lymphocytes is seen with an increased ratio of cytotoxic CD8+ T cells to T-regulatory cells.34,35 Select chemotherapeutic drugs have been demonstrated to be particularly potent inducers of the innate and adaptive antitumor immune systems and induce a distinct form apoptosis termed immunogenic cell death (ICD).36,37 Biochemical analysis has revealed distinctive features of ICD (in contrast to nonimmunogenic cell death); these include the expression of calreticulin at the cell surface as well as secretion of adenosine triphosphate during the apoptotic process.38 Several chemotherapeutic agents that appear to be effective at inducing ICD are cyclophosphamide, doxorubicin, and oxaliplatin39 ; relevantly, cyclophosphamide and doxorubicin are key components of the CHOP backbone used in the front-line management of PTCL. Chemotherapy that induces ICD has increased efficacy when administered to immune-competent mice using syngeneic models relative to when the tumors are grown in T-cell–deficient mouse models.38 Anthracycline-killed tumor cells are particularly effective at triggering an antitumor immune response,40 and have thus been used to create therapeutic cancer vaccines.41 Aside from inducing ICD, etoposide, cyclophosphamide, and vincristine (all components of the CHOEP and DA-EPOCH regimens) increase antigen presentation and MHC expression, thus triggering an antitumor immune response.37,42 Gemcitabine, a drug with substantial single-agent activity against TCLs,43,44 suppresses the amount of circulating myeloid-derived suppressor cells, favors polarization of macrophages toward the proinflammatory M1 phenotype, and increases antigenicity of tumor cells via the upregulation of MHC expression.45 Because chemotherapeutic agents can accentuate the antitumor immune response via pleiotropic effects on the TME, there is great interest in exploring the efficacy of these therapies in combination with drugs that stimulate T-cell immunity, such as checkpoint inhibitors.
Checkpoint inhibitors in TCLs
In TCLs, PD-1 and programmed death ligand 1 (PD-L1) expression on tumor cells and nonmalignant bystander T cells is frequent. Ninety-three percent of angioimmunoblastic T-cell lymphomas (AITLs) and 62% of PTCL-NOS have increased numbers of extrafollicular PD-1+ cells, contributing to an immune-suppressive microenvironment.46 In a large series of TCL patients (N = 155),47 PD-L1 was expressed by lymphoma cells in 27% of CTCL and 15% of PTCL biopsy samples. PD-L1 expression within the TME was more common, being observed in 73% and 39% of CTCL and PTCL cases, respectively. Given the frequent expression of PD-1 and PD-L1 in TCL, whereby expression may be prognostic or correlate with stage,48-50 antibodies targeting these proteins are being explored preclinically and clinically.
In addition to PD-1/PD-L1 expression, tumor mutational burden (TMB) is a predictive factor associated with response to immune CPB.51 The mutational profile of human malignancies has been extensively profiled,52 and tumors with increased mutational burdens, such as melanoma and non–small cell lung cancer, have improved responsiveness to immune CPB. Conversely, tumors associated with low-level mutational burden (acute lymphoblastic leukemia, acute myeloid leukemia) tend to be resistant. Responsiveness of patients with high TMB to CPB is thought to be secondary to expression of neoantigens (mutated antigens expressed in tumor and not normal cells) capable of stimulating an antitumor immune response.
In PTCL, mutational load varies significantly by subtype53 and in some cases approaches that observed in melanoma.54 Patients with PTCL-NOS and a p53 mutation have a significantly higher TMB relative to PTCL-NOS patients without a p53 mutation.55 Although not systematically reported, the mutational load among patients with PTCL likely varies by subtype, histology, and disease stage.
Nonsynonymous point mutations occur in CTCL at a rate of ≈3 mutations per megabase, a rate that is substantially higher than many other hematologic malignancies and also is higher than CPB-responsive solid tumors such as clear cell kidney cancer56 and breast cancer.54,57 Next-generation sequencing studies highlight recurrent mutational events in CTCL, which comprise focal deletions/amplifications, fusion events, and larger genomic rearrangements.58,59 Considerable variability has been reported, with particularly high mutation rates in cases that have undergone large cell transformation.60,61
PD-1/PD-L1 antibody therapy may promote malignant T-cell proliferation
In TCLs, where the T cells are malignant, the T-cell activation that results from checkpoint inhibition may have unintended and paradoxical consequences. Using a preclinical PTCL mouse model, investigators sought to explore the effects of PD-1/PD-L1 pathway inhibition on the TME.62 Wartewig et al62 created a novel PTCL model that relied on translocation of ITK-SYK, which had been observed in 5 of 46 PTCL cases.63 A single pulse of cre was sufficient to induce expansion of ITK–SYKCD4-CreERT2 lymphocytes; however, this proliferation was short-lived secondary to cell-intrinsic tumor-suppressive mechanisms. A subsequent whole-genome screen using a piggybac transposition system identified PDCD1, the gene encoding PD-1, as a potential tumor suppressor. Genetic and pharmacologic knockdown of PD-1 led to sustained proliferation of ITK–SYKCD4-CreERT2 lymphocytes, confirming that PD-1 functions as a tumor suppressor in this system. A meta-analysis of TCL patients demonstrated that 36 of 158 patients had a PDCD1 mutation, supporting a pathogenic role for PD-1 in TCL.62
A case report was subsequently published of a patient treated with PD-1 antibody therapy for an epithelial neoplasm that developed a secondary TCL. The investigators analyzed biopsy samples before and after checkpoint inhibition through TCR sequencing. A monoclonal T-cell population was present in <1% in the initial lung biopsy, which proliferated after 4 cycles of pembrolizumab therapy, ultimately manifesting as an aggressive PTCL-NOS. The authors concluded that the development or progression of this PTCL-NOS was secondary to pembrolizumab treatment. The US Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) database was reviewed and 12 similar cases of TCL originating after PD-1 antibody treatment have been reported. These findings provide additional evidence that PD-1 may function as a tumor suppressor of malignant T-cell clones.64
Clinical trials of PD-1 antibody therapy in TCL
A clinical trial of PD-1 immunotherapy for human T-cell leukemia-lymphoma virus (HTLV)–associated adult T-cell leukemia lymphoma (ATLL) was stopped prematurely due to rapid progression of the initial enrolled patients,65 a finding that appeared to validate Wartewig et al’s preclinical findings. Peripheral blood from 2 of the 3 patients experiencing rapid progression was analyzed before and after treatment with nivolumab. Genomic DNA isolated from the peripheral blood monocytic cells (PBMCs) demonstrated a rapid expansion of the malignant clones after a single infusion of PD-1 antibody therapy. The malignant ATLL subclones demonstrated a phenotype characteristic of T-regulatory cells, expressing genes such as MAGEH1, CCR8, CTLA4, CD27, CD70, TNFRSF4, and LAG-3.66
A similar clinical trial of TCL-leukemia patients was performed in Japan, and of the 8 patients enrolled at the time of the report, none experienced evidence of rapid progression of their disease.67 Given the small numbers of patients enrolled in both trials, it is difficult to speculate regarding potential etiologies of the discrepant results in the American65 and Japanese67 trials. However, the obvious difference between the trials was that the American trial enrolled patients with a relatively indolent course prior to study enrollment; 2 patients had slow disease progression on prior therapy, and 1 patient had stable disease. Conversely, the Japanese trial exclusively enrolled patients with aggressive ATLL with adverse prognostic factors. Along these lines, Rauch et al worked to develop a mouse model of hyperprogressive disease in aggressive ATLL; in all models evaluated, PD-1 blockade resulted in decreased tumor burden.66 To reliably determine whether indolent biology predisposes to hyperprogressive disease in ATLL patients treated with CPB, further study is necessary. An improved understanding of the tumor-suppressive role of PD-1 in indolent vs aggressive ATLL may allow for the development of predictive biomarkers to CPB.
Other early-phase clinical trials of immunotherapy in PTCL patients have yielded mixed results. In phase 1/2 studies, pretreated PTCL patients had a response rate of ∼30% to nivolumab and pembrolizumab.13,68-70 Duration of response is more limited than what has been observed in solid tumor malignancies and is ∼3 months.13 PD-1 antibody therapy leads to a reduction in the number of circulating T-regulatory cells as a percentage of total CD4+ T cells. Increased percentages of circulating CD4+ T cells pretreatment are associated with improved responses to checkpoint-inhibitor therapy.13 Given the modest efficacy of PD-1/PD-L1 antibodies as a single agent in PTCL, studies are investigating combination approaches that may overcome CPB-resistance mechanisms (described in "Rational combinations involving CPB in TCLs").
Checkpoint inhibitors appear to be a particularly encouraging strategy in Epstein-Barr virus (EBV)–associated lymphoproliferative disorders.71-74 Viral-associated malignancies have decreased receptor diversity as well as alterations in the tumor-immune microenvironment that may modulate sensitivity to immune CPB.75 Viruses can incorporate into the malignant genome, leading to increased expression of PD-L1 or PD-L2.76 Extranodal natural killer (NK)–cell lymphomas are uniformly associated with infection by EBV. In 1 report, 5 of 7 patients with relapsed/refractory NK/TCL after l-asparaginase–based therapy had a complete response to pembrolizumab (2 partial responses).73
The efficacy of PD-1/PD-L1 inhibition is also being evaluated in CTCL patients. In a heavily pretreated advanced-stage cutaneous TCL cohort of 24 patients (with MF or SS), pembrolizumab showed an overall response rate of 38% with a median duration of response not reached.69 Interestingly, 53% of patients with SS experienced a transient worsening of cutaneous symptoms (ie, worsening erythroderma and pruritus). This flare reaction correlated with PD-1 expression on Sézary cells and did not result in treatment discontinuation in any patient. There was no correlation between PD-L1 expression by immunohistochemistry and response rate. Similarly, an 18-gene interferon γ gene-expression score was not increased in responders (in contrast to what has been observed in solid tumors77 ). A nonsignificant trend was observed that patients responding to pembrolizumab had increased total nonsynonymous mutations within their biopsy specimen.69
A potential explanation of the variable observations seen when evaluating the role of PD-1/PD-L1 blockade in the treatment of TCLs is the complexity of the effects of CPB on the tumor-immune microenvironment. When cultured with leukemic cells, NK cells demonstrate increased expression of PD-L1 via an AKT-dependent mechanism. PD-L1 antibody therapy leads to activation of PD-L1+ NK cells, an effect that may contribute to the efficacy of PD-L1 antibody therapy in tumors that do not express PD-L1.78 Similarly, PD-1, in addition to being expressed on T cells, is also frequently expressed on B cells. PD-1 expression on B-cell precursors tends to be minimal, and expression increases with B-cell maturation. Upon activation by the presence of Toll-like receptor 9, blockade of the PD-1/PD-L1 axis leads to increased B-cell activation, proliferation, and release of proinflammatory cytokines.79 Thus, the immune modulation of the TME that results from treatment with PD-1/PD-L1 antibodies in TCLs is complex and only partially explained by direct effects on the malignant T cell.
Targeting PD-1 vs PD-L1 in TCL
Because PD-1, unlike PD-L1, is frequently expressed on the normal T-cell population, it is worth considering whether treatment with PD-1 and PD-L1 antibody therapy will have distinct effects in TCL. Treatment with PD-1 antibody therapy inhibits PD-1 interaction with both PD-L1 and PD-L2 on the tumor cells. PD-L2 has been demonstrated to bind PD-1 with twofold to sixfold higher affinity relative to PD-L1.80 However, constitutive basal expression of PD-L2 tends to be lower than PD-L1.81 Although the role of PD-L2 in creating an immune-suppressive tumor-immune microenvironment is incompletely understood, data are emerging that PD-L2 expression on tumor cells (alone and in combination with PD-L1) functions to suppress the antitumor immune response of CD8+ T cells. Furthermore, PD-L2 expression on tumor cells has been shown to mediate resistance to treatment with PD-L1 antibody therapy; this resistance is overcome by treatment with PD-L2 or PD-1 antibody therapy.82 Nevertheless, in at least 1 preclinical model of PTCL, PD-1 and PD-L1 antibody therapy were both identified as haploinsufficient tumor suppressors of lymphomagenesis.62 Further studies are necessary to more fully understand the use of PD-1 vs PD-L1 antibody therapy in the treatment of TCLs.
Non–PD-1/PD-L1 checkpoints in TCL
In addition to PD-1/PD-L1, other checkpoint molecules may play a role in T-cell lymphomagenesis.
T-cell immunoreceptor with Ig and ITIM domains (TIGIT) functions as a coinhibitory receptor and downregulates Th1 and Th17 differentiation of T cells while promoting Th2 differentiation. TIGIT+ T cells are seen with increased frequency in cutaneous plaques of patients with MF and SS. Patients with SS have a 34% increase in the prevalence of circulating TIGIT+ CD4 T cells relative to healthy controls.83,84 LAG-3 is a coinhibitory receptor that structurally resembles the CD4 molecule and binds MHC II with increased affinity relative to CD4.85 LAG-3 serves as a checkpoint molecule that inhibits the antitumor T-cell response and has a protective effect against FS7-associated cell surface antigen (FAS)–mediated apoptosis.86 LAG-3, in contrast to what is observed with TIGIT, is downregulated in circulating CD4 T cells in patients with MF and SS.50,87 The etiopathogenic role of LAG-3 and TIGIT in MF and SS has yet to be fully elucidated.
The CTLA-4/CD28/B7 axis is recurrently altered in TCLs. A CTLA4-CD28 fusion gene, which codes for the extracellular domain of CTLA-4 and the intracellular signaling domain of CD28, was identified using RNA sequencing in a patient with AITL. Subsequent Sanger sequencing in 115 cases of TCLs identified this fusion in 58% of AITL cases, 23% of PTCL-NOS cases, and 29% of NK/TCLs.88 Although the exact frequency of CTLA4-CD28 fusion is controversial,89 given the key role played by the CTLA4/CD28/B7 axis in T-cell proliferation, it logically follows that aberrant regulation of this pathway can be pathogenic in the development of TCLs. Recurrent activating mutations in CD28 have also been identified in patients with PTCL, resulting in increased transcription of CD28-responsive genes CD226 and TNFA.90 In the most common type of CTCL, MF, there is increased CTLA-4 expression in PBMCs relative to control patients, and higher expression of CTLA-4 was correlated with more advanced cases of MF.90,91
There is anecdotal evidence that alterations in the CTLA4-CD28 axis may be targetable in patients with CTCL. A patient with SS was identified to have a CD28-CTLA4 fusion using whole-genome sequencing. The patient was treated with ipilimumab and experienced a profound disease response, including 50% reduction in erythema and 75% reduction in size of dermal tumors.92 This case report suggests that further study of the use of a personalized approach in PTCL patients with genetic alterations in CTLA4 and/or CD28 is warranted.
CD47 is expressed on all normal cells and serves as the principal ligand for signal regulatory protein α (SIRPα), expressed on phagocytic cells. CD47 functions as an antiphagocytic “don’t eat me” signal, and thus can be considered a checkpoint molecule within the innate immune system. CD47 is often overexpressed on cancer cells in order to evade immune surveillance, and increased CD47 expression is correlated with adverse outcomes in various subtypes of NHL.93 HuF59 is a humanized monoclonal antibody against CD47 and was shown to synergize with rituximab in the management of a variety of preclinical models of NHL.94 Subsequently, a phase 1 clinical trial of Hu5F9 in combination with rituximab was performed in patients with NHL (several subtypes). Of 22 evaluable patients, 11 had an objective response to the therapy. Responses were durable, with 91% of responses ongoing at time of data cutoff for publication.95 TTI-621 is a separate drug in clinical trials that is designed to target CD47. TTI-621 is a recombinant fusion protein that contains the CD47-binding domain of human SIRPα N-terminal domain fused to the Fc receptor of immunoglobulin G1 (IgG1). Due to the presence of IgG1, a strong antibody-dependent cellular cytotoxicity component is triggered by TTI-621.96 In a preliminary report of a phase 1 clinical study, it was demonstrated that TTI-621 led to a decrease in the quantity of the dominant malignant clone in the peripheral blood in 4 of 5 patients with SS. Lactate dehydrogenase, which can be used as a tumor marker in patients with SS, showed marked improvement after a single dose of TTI-621. The treated patients also had a corresponding decrease in their erythroderma.97
Rational combinations involving CPB in TCLs
Several approaches involving rational combinations of checkpoint antibodies combined with other immune-modulating therapies are being explored (Table 1). There is extensive solid tumor experience using the combination of PD-1/PD-L1 antibodies with chemotherapy.98 Preclinical evidence supporting such an approach comes from studies demonstrating that immunogenic chemotherapy may sensitize malignancies to CPB by promoting T-cell recognition and depleting immune-suppressive cells in the TME99 (Figure 1). As detailed in "PD-1/PD-L1 antibody therapy may promote malignant T-cell proliferation," in some TCL patients, checkpoint inhibition may lead to rapid proliferation of the malignant clone; this effect may theoretically be exploited therapeutically by treating with concurrent cytotoxic chemotherapy. The chemotherapy would be expected to have increased efficacy in rapidly dividing malignant T cells. A currently accruing clinical trial, in which first-line standard-of-care chemotherapy (ie, EPOCH) is combined with nivolumab in PTCL patients (NCT03586999), will help evaluate this hypothesis.
Ongoing trials of PD-1/PD-L1 inhibition in TCL
| Study name | Study no. | Study phase |
|---|---|---|
| Study of pembrolizumab in patients with early-stage NK/T-cell Lymphoma, nasal type | NCT03728972 | 2 |
| Pembrolizumab and pralatrexate in treating patients with relapsed or refractory peripheral T-cell lymphomas | NCT03598998 | 1/2 |
| Pembrolizumab in relapsed or refractory extranodal NK/T-cell lymphoma, nasal type and EBV-associated diffuse large B-cell lymphomas | NCT03586024 | 1/2 |
| A trial assessing the effect of pembrolizumab combined with radiotherapy in patients with relapsed, refractory, specified stages of cutaneous T-cell lymphoma (CTCL) mycosis fungoides (MF)/Sézary syndrome (SS) (PORT) | NCT03385226 | 2 |
| Study of pembrolizumab (MK-3475) in combination with romidepsin | NCT03278782 | 1/2 |
| Study of pembrolizumab combined with decitabine and pralatrexate in PTCL and CTCL | NCT03240211 | 1 |
| Pembrolizumab and external beam radiation therapy in treating patients with relapsed or refractory non-Hodgkin lymphoma | NCT03210662 | 2 |
| Phase 2 trial of nivolumab for pediatric and adult relapsing/refractory ALK+ anaplastic large cell lymphoma, for evaluation of response in patients with progressive disease (cohort 1) or as consolidative immunotherapy in patients in complete remission after relapse (cohort 2) (NIVO-ALCL) | NCT03703050 | 2 |
| Nivolumab with standard of care chemotherapy for peripheral T-cell lymphomas | NCT03586999 | 1/2 |
| Durvalumab in different combinations with pralatrexate, romidepsin and oral 5-azacitidine for lymphoma | NCT03161223 | 1/2 |
| Durvalumab with or without lenalidomide in treating patients with relapsed or refractory cutaneous or peripheral T-cell lymphoma | NCT03011814 | 1/2 |
| PARCT: Trial of atezolizumab in relapsed/refractory cutaneous T-cell lymphoma (CTCL) (PARCT) | NCT03357224 | 2 |
| Avelumab in relapsed and refractory peripheral T-cell lymphoma (AVAIL-T) | NCT03046953 | 2 |
| Study name | Study no. | Study phase |
|---|---|---|
| Study of pembrolizumab in patients with early-stage NK/T-cell Lymphoma, nasal type | NCT03728972 | 2 |
| Pembrolizumab and pralatrexate in treating patients with relapsed or refractory peripheral T-cell lymphomas | NCT03598998 | 1/2 |
| Pembrolizumab in relapsed or refractory extranodal NK/T-cell lymphoma, nasal type and EBV-associated diffuse large B-cell lymphomas | NCT03586024 | 1/2 |
| A trial assessing the effect of pembrolizumab combined with radiotherapy in patients with relapsed, refractory, specified stages of cutaneous T-cell lymphoma (CTCL) mycosis fungoides (MF)/Sézary syndrome (SS) (PORT) | NCT03385226 | 2 |
| Study of pembrolizumab (MK-3475) in combination with romidepsin | NCT03278782 | 1/2 |
| Study of pembrolizumab combined with decitabine and pralatrexate in PTCL and CTCL | NCT03240211 | 1 |
| Pembrolizumab and external beam radiation therapy in treating patients with relapsed or refractory non-Hodgkin lymphoma | NCT03210662 | 2 |
| Phase 2 trial of nivolumab for pediatric and adult relapsing/refractory ALK+ anaplastic large cell lymphoma, for evaluation of response in patients with progressive disease (cohort 1) or as consolidative immunotherapy in patients in complete remission after relapse (cohort 2) (NIVO-ALCL) | NCT03703050 | 2 |
| Nivolumab with standard of care chemotherapy for peripheral T-cell lymphomas | NCT03586999 | 1/2 |
| Durvalumab in different combinations with pralatrexate, romidepsin and oral 5-azacitidine for lymphoma | NCT03161223 | 1/2 |
| Durvalumab with or without lenalidomide in treating patients with relapsed or refractory cutaneous or peripheral T-cell lymphoma | NCT03011814 | 1/2 |
| PARCT: Trial of atezolizumab in relapsed/refractory cutaneous T-cell lymphoma (CTCL) (PARCT) | NCT03357224 | 2 |
| Avelumab in relapsed and refractory peripheral T-cell lymphoma (AVAIL-T) | NCT03046953 | 2 |
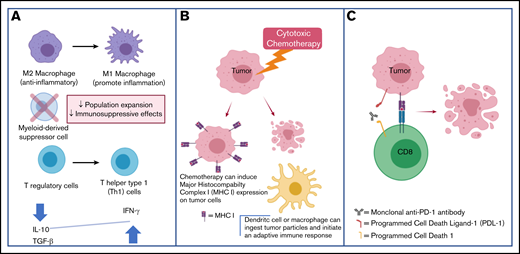
Cytotoxic chemotherapy plus CPB may be synergistic through the following mechanisms. (A) Chemotherapy can reverse the immune-suppressive TME. (B) Chemotherapy can increase tumor antigen presentation and facilitate an immune response. (C) CPB promotes antitumor T-cell responses.
Cytotoxic chemotherapy plus CPB may be synergistic through the following mechanisms. (A) Chemotherapy can reverse the immune-suppressive TME. (B) Chemotherapy can increase tumor antigen presentation and facilitate an immune response. (C) CPB promotes antitumor T-cell responses.
Evidence has emerged that expression of MHC II, a molecule involved in antigen presentation, correlates with response to PD-1/PD-L1 antibody therapy.100 Although tumors generally express MHC I on their surface, expression of MHC II is more variable. MHC II is classically expressed on APCs, though there is increasing recognition of MHC II on the surface of other cell types as well, including epithelial cells.101 The MHC II molecule is responsible for presenting antigens to CD4+ T cells, a key step in the inflammatory response.102 In cutaneous TCL, it has been demonstrated that MHC II expression is enhanced in the tumor-involved areas, an effect that likely contributes to an inflamed TME.103 One therapeutic approach that has emerged to increase MHC expression in malignancies, as demonstrated by our group and others, is via the use histone deacetylase inhibitors (HDACi’s).104-107 As a result, there is extensive preclinical108 and clinical (NCT03765229) evaluation ongoing of combined therapy with HDACi’s and PD-1/PD-L1 CPB.109 Such a combinatorial approach may be particularly effective in TCL given that single-agent treatment with HDACi’s is an established and efficacious therapeutic approach in this disease type. Preliminary results of a clinical trial evaluating pembrolizumab in combination with romidepsin in PTCL patients demonstrate a response rate of 44% (n = 15).110 The 3 patients who achieved a complete response remain disease-free >10 months at time of latest follow-up.
Hypomethylating agents remodel the TME, increasing lymphocyte infiltration and expression of inflammatory cytokines leading to synergism with PD-1 antibody therapy in multiple tumor types.111-113 5-azacytidine has single-agent activity in patients with AITL, inducing an objective response in 6 of 12 patients (50%) in a retrospective case series.114 A phase 1 dose-escalation trial evaluated 5-azacytidine in combination with romidepsin for the management of patients with malignant lymphomas; 11 of the 31 enrolled patients had PTCL. The objective response rate was 10% in patients with non-PTCL and 73% among patients with PTCL.115 These results suggest that combined epigenetic modification therapy may be potently active in patients with PTCL. This regimen is also being explored in combination with immunotherapy. The DURABILITY trial is investigating, among other combinations, triplet treatment with durvalumab, 5-azacytidine, and romidepsin in patients with PTCL (NCT0316223). Preliminary results show complete responses in all 3 evaluable patients assigned to this treatment to date and more mature results are eagerly awaited.116
Chimeric antigen receptor (CAR) T cells have become increasingly used for the management of patients with hematologic malignancies. The development of CAR T cells to treat patients with PTCL has proved challenging due to concerns of fratricide as well as severe immunosuppression associated with depletion of normal T cells. However, CAR T cells directed against T-cell antigens CD5 and CD7 have shown promise in preclinical studies of PTCL.117,118 In a clinical trial evaluating CD5-directed CAR T therapy, 4 of 9 treated patients had an objective response and there were no instances of grade 3/4 cytokine release syndrome or neurotoxicity.119 Checkpoint inhibitors are being evaluated in combination with CAR T cells in an effort to mitigate immune exhaustion of the transplanted T cells. Repeated antigen exposure by CAR T cells leads to T-cell senescence and expression of inhibitory receptors, PD-1, CTLA4, TIGIT, LAG-3, CD244, CD160, and TIM3.120 Clinical trials evaluating combination therapy of CAR T therapy with CPB suggest the combination is safe118 ; PD-1 antibody therapy may stimulate expansion of the CAR T-cell population leading to synergistic antilymphoma efficacy.121 Evaluation of combined CPB with CAR T therapy for PTCL has been limited by concerns that PD-1 antibody therapy will stimulate proliferation of the malignant T-cell population instead of (or in addition to) the CAR T cells. No clinical trials of combined CPB and CAR T therapy have been published to date in PTCL. Identification of predictive factors in TCL for response to CPB may permit the use of PD-1 antibodies to selectively induce expansion of CAR T cells while simultaneously having anti-PTCL activity in carefully selected patients.
Conclusion
Historically, chemotherapy has formed a backbone of therapy for patients with TCLs. However, in recent years, with the success of checkpoint inhibitors in multiple solid and hematologic malignancies, cancer researchers have developed a profound interest in evaluating means of optimizing antitumor immunity within the TME. The TME in TCLs exhibits an immune-suppressive phenotype that blunts the antitumor immune response. Preliminary evidence suggests that checkpoint inhibitors, such as PD-1 antibody therapy, may remodel the TME to promote the infiltration of activated immune cells that contribute to an antitumor immune response. Additional studies are under way to evaluate synergistic combinations with CPB, such as combining PD-1 antibody therapy with immune-modulating therapies including immunogenic chemotherapy and epigenetic modifiers. Conversely, some preclinical and clinical evidence suggests that PD-1 antibody therapy may actually accelerate TCL progression. The efficacy of checkpoint inhibition in TCL appears to be model (in preclinical studies) and patient (in clinical studies) dependent. The current review is designed to serve as a “call to action” to better understand the immune landscape of TCL in order to facilitate identification of predictive biomarkers for favorable response to PD-1 antibody therapy. In the absence of availability of established biomarkers, it is imperative that clinical trials evaluating CPB in TCL be based on sound scientific rationale and that they incorporate stringent study oversight and interim analysis to minimize potential harm to enrolled subjects. Taken together, there is exciting progress being made in understanding the role of immune dysfunction in TCL progression, and it appears inevitable that immune-modulating therapies will assume an increasing role in the management of TCL patients.
Data sharing requests may be e-mailed to the corresponding author, Bradley Haverkos, at bradley.haverkos@cuanschutz.edu.
Authorship
Contribution: A.N. was a primary author; T.A.-J. and E.D. helped with writing and editorial support; and B.H. developed the idea and was a primary author.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bradley Haverkos, University of Colorado, 1665 Aurora Ct F754, Aurora, CO 80045; e-mail: bradley.haverkos@cuanschutz.edu.

