

Key Points
Ponatinib produces a coronary microangiopathy that mimics myocardial infarction and can be detected rapidly by contrast echocardiography.
N-acetylcysteine therapy can potentially resolve ischemic complications caused by ponatinib-related microangiopathy.
Introduction
The third-generation receptor tyrosine kinase-inhibitor (TKI) ponatinib is used for the treatment of chronic myelogenous leukemia (CML) and Philadelphia chromosome–positive acute lymphoblastic leukemia. It is most commonly prescribed when drug resistance is produced by the T315I mutation in the BCR-ABL kinase domain.1 This TKI is associated with a particularly high incidence of adverse events involving acute arterial occlusion that can manifest as stroke and myocardial injury or infarction.2,3 In those with myocardial infarction, there is a paucity of information on the prevalence having a culprit critical obstruction on coronary angiography. While the pathobiology of thrombotic events in humans is unknown, preclinical studies in mice suggest that high-dose ponatinib therapy can produce von Willebrand factor (VWF)–mediated platelet-endothelial adhesion, which results in a microvascular angiopathy and regional ischemic myocardial dysfunction.4 We present a case of ponatinib-related coronary microvascular angiopathy that highlights new approaches for noninvasive diagnosis and treatment of the condition that were translated from preclinical experience.
Case description
A 30-year-old male with CML presented to the hospital with fever, malaise, leukocytosis, and chest discomfort for several days. He had been diagnosed with CML 10 years prior to presentation. He was initially treated with imatinib and then dasatinib for kinase-independent resistance. He was then switched to ponatinib 2 years prior to presentation for increase in disease activity (elevated white blood cell [WBC] count) and evidence for the T315I mutation in 100% of cells. His dose of ponatinib was 30 to 45 mg/day. He was also on hydroxyurea for several months prior to presentation based on disease activity (elevated WBC count) after the ponatinib dose was reduced to 30 mg/day for portal vein thrombosis. His only atherosclerotic risk factor was former tobacco smoking (8 pack-year history). A recent lipid profile was normal. On presentation, a 12-lead electrocardiogram (ECG) showed ST elevation of >3 mm in the inferior and lateral leads (Figure 1C). Serum troponin T was elevated at 3.95 mg/dL (Figure 1A). Because of thrombocytopenia and elevated WBC count (platelet count 25×103/mm3, white blood cell count 181×103/mm3), emergent coronary angiography was deferred. He was started on a β blocker, hydroxyurea was continued, and ponatinib was discontinued. Urgent leukapheresis was performed, but failed to improve his symptoms or ST elevation on ECG. Transthoracic echocardiography in the intensive care unit detected multiple segmental regions of akinesis that were somewhat atypical for conventional coronary artery disease.
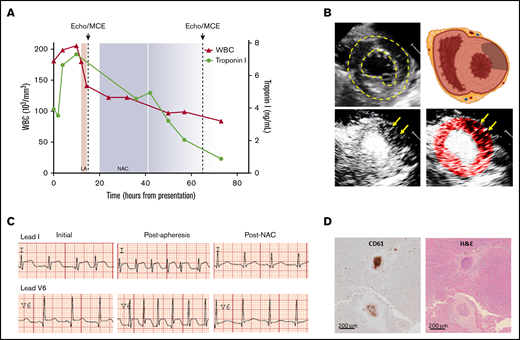
Clinical and imaging data highlighting manifestations of ponatinib-mediated coronary thrombotic microangiopathy. (A) Temporal trends for WBC count (red triangles) and serum troponin I (green circles) relative to the timing of leukapheresis (LA), echocardiography with MCE (dashed lines), and initiation of NAC (shaded area extending to 2 half-lives after last dose). (B) Transthoracic echocardiography demonstrating left ventricular endocardial and epicardial borders (dashed lines) in the midventricular parasternal short-axis plane on noncontrast 2-dimensional imaging and a schematic showing cardiac orientation with gray region overlying the anterolateral region where perfusion was abnormal (upper panels) and corresponding MCE perfusion imaging (bottom panels) prior to NAC treatment with a tissue-subtraction algorithm whereby opacification occurs only where microbubble contrast agent is present, demonstrating patchy regions lacking perfusion (arrows) in the anterolateral region. The bright central region represents the opacified ventricular cavity. The color-coded image is provided to highlight regions lacking perfusion within the confines of the left ventricular borders. (C) Lead I and V6 tracings from ECGs performed on initial presentation, after leukapheresis and approximately 24 hours after initiation of NAC (see supplemental Figure 1 for the full 12-lead ECG on presentation). (D) Postmortem immunohistochemistry for platelet CD61 (β3-integrin) and corresponding hematoxylin and eosin staining showing evidence recent platelet-rich microvascular thrombus and mural hyperplasia involving arterioles. Brown indicates secondary peroxidase staining.
Clinical and imaging data highlighting manifestations of ponatinib-mediated coronary thrombotic microangiopathy. (A) Temporal trends for WBC count (red triangles) and serum troponin I (green circles) relative to the timing of leukapheresis (LA), echocardiography with MCE (dashed lines), and initiation of NAC (shaded area extending to 2 half-lives after last dose). (B) Transthoracic echocardiography demonstrating left ventricular endocardial and epicardial borders (dashed lines) in the midventricular parasternal short-axis plane on noncontrast 2-dimensional imaging and a schematic showing cardiac orientation with gray region overlying the anterolateral region where perfusion was abnormal (upper panels) and corresponding MCE perfusion imaging (bottom panels) prior to NAC treatment with a tissue-subtraction algorithm whereby opacification occurs only where microbubble contrast agent is present, demonstrating patchy regions lacking perfusion (arrows) in the anterolateral region. The bright central region represents the opacified ventricular cavity. The color-coded image is provided to highlight regions lacking perfusion within the confines of the left ventricular borders. (C) Lead I and V6 tracings from ECGs performed on initial presentation, after leukapheresis and approximately 24 hours after initiation of NAC (see supplemental Figure 1 for the full 12-lead ECG on presentation). (D) Postmortem immunohistochemistry for platelet CD61 (β3-integrin) and corresponding hematoxylin and eosin staining showing evidence recent platelet-rich microvascular thrombus and mural hyperplasia involving arterioles. Brown indicates secondary peroxidase staining.
Methods
Immediately following functional assessment with transthoracic echocardiography, bedside microvascular perfusion imaging with myocardial contrast echocardiography (MCE) was performed. This method relies on the acoustic signal produced by microbubbles as they transit through the coronary microcirculation and provides key information on perfusion, including the blood microvascular flux rate and functional patency of the microcirculation (microvascular blood volume).5,6 MCE was performed using a continuous infusion of Definity (1 vial in 30 mL, 1.0-1.5 mL/min) and low-power multipulse contrast-specific imaging (Power Modulation imaging, iE33; Philips Ultrasound, Andover, MA).5
Results and discussion
MCE was immediately interpreted at the bedside and thought to reveal an unusual pattern of heterogeneous patches where microvascular perfusion was absent in the same regions with wall motion abnormality (Figure 1B). These findings are aligned with those described in mice with VWF-mediated microangiopathy after ponatinib treatment.4 Because N-acetylcysteine (NAC) can inhibit VWF self-association7 and was recently shown to prevent ponatinib-related microvascular angiopathy in mice,4 the patient was treated with IV NAC using a 150 mg/kg load, followed by 50 mg/kg twice 12 hours apart. Within 24 hours of NAC treatment, the patient’s symptoms nearly completely resolved, ST segments normalized, and troponin I levels decreased (Figure 1A). Repeat MCE after 48 hours of initiating NAC demonstrated near-complete resolution of microvascular perfusion but persistent systolic dysfunction. The patient was discharged several days later with a plan for outpatient coronary angiography and re-evaluation of ventricular function. Unfortunately, 2 weeks after discharge, he died of noncardiovascular complications, primarily overwhelming sepsis from an existing wound infection. On autopsy, there was no evidence of epicardial coronary artery atherosclerosis, but CD61-positive straining thrombotic occlusion of multiple intramyocardial small arteries and large arterioles was found and believed to be several weeks old based on surrounding hyperplasia (Figure 1D), consistent with a thrombotic microvascular angiopathy.
Oxidative modification of VWF and ADAMTS13, the enzyme that cleaves VWF, has been implicated in platelet-endothelial adhesion in diseases such as atherosclerosis and acquired thrombotic thrombocytopenic purpura,8,9 and may be the culprit in the adverse prothrombotic events linked to ponatinib therapy. In mice treated with ponatinib, in vivo molecular imaging of vascular phenotype has demonstrated increased vascular-endothelial–associated VWF in its active conformation and secondary endothelial adhesion of platelets.4 The presence of platelet adhesion in the coronary microcirculation was associated with echocardiographic wall motion abnormalities. All of these findings tended to be worse in mice with hyperlipidemia and atherosclerosis produced by gene-targeted deletion of apolipoprotein E and high-fat diet. NAC was found to largely prevent these adverse events. Currently, studies using preclinical models of thrombotic thrombocytopenic purpura suggest that prophylactic administration of NAC can be effective in preventing severe disease by decreasing VWF multimer size, but efficacy is lower for its use to resolve disease in progress once thrombi develop.10 Several case reports have demonstrated effective use of IV NAC administration as an adjunct to therapy in patients with thrombotic thrombocytopenic purpura with evidence of clinical benefit.11,12
We believe that MCE is an ideal approach for assessing TKI-related complications for several reasons. It can be performed rapidly at the bedside, provides information immediately to the clinician, and yields spatial information on functional patency of the microcirculation that becomes abnormal in microvascular thrombotic disease.13 As this case illustrates, the ability to examine perfusion also allows clinicians to temporally assess response to therapies designed to reverse microvascular obstruction. Improvements in regional left ventricular motion are not as useful for monitoring response because of myocardial stunning whereby wall motion abnormalities persist for a period of time after resolution of ischemia.
This case is the first described use of bedside MCE for diagnosis of TKI-related microvascular angiopathy. It also suggests that ischemic complications caused by ponatinib-related microangiopathy can be resolved by NAC. Our finding in this patient opens new opportunities for point-of-care diagnosis and treatment monitoring of thrombotic angiopathies.
For data sharing requests, e-mail the corresponding author, Jonathan R. Lindner (lindnerj@ohsu.edu).
Acknowledgments
J.R.L. is supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01-HL078610, R01-HL130046, and P51-OD011092; and National Aeronautics and Space Administration grant 18-18HCFBP_2-0009. M. D. Wu is supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant K08-HL133493.
Authorship
Contribution: M. D. Wu and J.R.L. wrote the case report; J.R.L. and J.H. performed and interpreted MCE data; B.M., K.K., and S.O. contributed clinical data to the manuscript and participated in the experimental administration of NAC; and A.G. and M. D. Wood performed histology and immunohistochemistry specific to microvascular angiopathies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jonathan R. Lindner, Knight Cardiovascular Institute, UHN-62, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Portland, OR 97239; e-mail:lindnerj@ohsu.edu.

