

Key Points
Murine TRALI antibody pathogenicity is not associated with Fab- or Fc-binding affinities nor with the composition of Fc glycans.
Murine TRALI antibody exhibits increased Fc-mediated complement activation, which does not significantly depend on Fc glycosylation.

Abstract
Transfusion-related acute lung injury (TRALI) remains a leading cause of transfusion-related deaths. In most cases, anti-leukocyte antibodies in the transfusion product trigger TRALI, but not all anti-leukocyte antibodies cause TRALI. It has been shown that the anti–major histocompatibility complex (MHC) class I antibody 34-1-2S (anti–H-2Kd) causes TRALI in BALB/c mice (MHC class I haplotype H-2Kd), whereas SF1.1.10 (anti–H-2Kd) does not. In C57BL/6 mice (MHC class I haplotype H-2Kb), TRALI only occurs when anti-MHC class I antibody AF6-88.5.5.3 (anti–H-2Kb) is administered together with a high dose of 34-1-2S. It remains unknown which specific antibody characteristics are responsible for eliciting TRALI. We therefore investigated several biological and structural features of 34-1-2S compared with other anti-MHC class I antibodies, which on their own do not cause TRALI: SF1.1.10 and AF6-88.5.5.3. No substantial differences were observed between the TRALI-causing 34-1-2S and the TRALI-resistant SF1.1.10 regarding binding affinity to H-2Kd. Regarding binding affinity to H-2Kb, only AF6-88.5.5.3 potently bound to H-2Kb, whereas 34-1-2S exhibited weak but significant cross-reactivity. Furthermore, the binding affinity to FcγRs as well as the Fc glycan composition seemed to be similar for all antibodies. Similar Fc glycosylation profiles were also observed for human TRALI-causing donor anti-HLA antibodies compared with human anti-HLA antibodies from control donors. 34-1-2S, however, displayed superior complement activation capacity, which was fully Fc dependent and not significantly dependent on Fc glycosylation. We conclude that TRALI induction is not correlated with Fab- and Fc-binding affinities for antigen and FcγRs, respectively, nor with the composition of Fc glycans; but increased Fc-mediated complement activation is correlated with TRALI induction.
Introduction
Administration of blood transfusions is a frequent and lifesaving intervention; however, life-threatening transfusion reactions may unexpectedly occur. This includes transfusion-related acute lung injury (TRALI), which remains one of the leading causes of transfusion-related fatalities.1 Upon blood transfusion, within 6 hours, acute respiratory distress occurs due to the development of increased endothelial permeability that leads to pulmonary edema resulting in TRALI. Apart from supportive measures (eg, oxygen), no therapeutic approaches are available for TRALI.1,2 The pathophysiology is complex and remains challenging to dissect. Generally, a 2-hit model is assumed to represent the framework of TRALI pathology, in which the first hit reflects the underlying clinical condition of the patient (eg, inflammation), and the second hit is conveyed by factors in the transfusion product.1 These factors in the transfusion product are, in the majority of the cases, anti-HLA class I/II antibodies or anti-human neutrophil antigen antibodies. In various types of murine TRALI models, the anti–major histocompatibility complex (MHC) class I antibody 34-1-2S (immunoglobulin G2a [IgG2a], anti–H-2Kd) is known to trigger TRALI.1 This antibody is sufficient to trigger TRALI in BALB/c mice (MHC class I haplotype H-2Kd),3-11 whereas other anti–H-2Kdantibodies of the same IgG2a subclass such as SF1.1.10 do not induce TRALI.7 Similarly, the H-2Kb–specific IgG2a antibody AF6-88.5.5.3 does not trigger TRALI in C57BL/6 (MHC class I haplotype H-2Kb) mice. However, when it is administered together with a high dose of 34-1-2S, TRALI is triggered, whereas 34-1-2S infusion alone does not induce TRALI (Figure 1).4,12,13 Earlier work suggested that intact 34-1-2S was required for murine TRALI induction since neither its F(ab)2 nor Fc fragments alone were able to mediate TRALI.14 However, which biological and structural properties of antibodies determine their pathogenic TRALI-inducing capability is currently unknown.
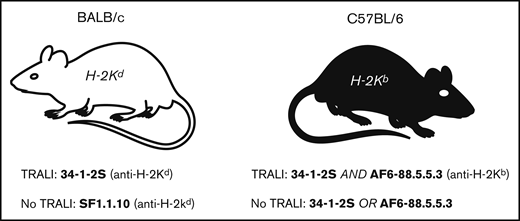
Overview of IgG2a anti-MHC class I antibodies that either cause TRALI or are unable to cause TRALI in mice. BALB/c (left) and C57BL/6 (right) mice are depicted. Notably, a higher dose of 34-1-2S, compared with AF6-88.5.5.3 (10:1), is required to obtain TRALI in C57BL/6 mice. Antibody-mediated TRALI induction or resistance is based on published literature, as indicated in the text.
Overview of IgG2a anti-MHC class I antibodies that either cause TRALI or are unable to cause TRALI in mice. BALB/c (left) and C57BL/6 (right) mice are depicted. Notably, a higher dose of 34-1-2S, compared with AF6-88.5.5.3 (10:1), is required to obtain TRALI in C57BL/6 mice. Antibody-mediated TRALI induction or resistance is based on published literature, as indicated in the text.
In the current article, the potential features that might explain the differences between TRALI-inducing and TRALI-resistant antibodies were studied. These include affinity to their cognate antigen, affinity to IgG-Fc receptors (FcγRs),15 and the ability to activate complement through binding of the first component of the classical complement pathway C1q.16 In addition, we examined the N-linked glycosylation of the IgG-Fc at position 297, for murine as well as human TRALI-causing antibodies, which may influence binding to FcγRs and/or activation of complement.17
Methods
Human TRALI and control donor plasma samples
The study used human TRALI donor plasma samples that were diagnostically found to contain anti-HLA antibodies with known specificity which caused a clinically confirmed TRALI reaction upon transfusion. The samples were obtained from Sanquin Diagnostics (Amsterdam, The Netherlands). Control donor plasma samples from women aged 40 to 59 years containing anti-HLA antibodies with known specificity were obtained from Sanquin Blood Supply (Amsterdam, The Netherlands).
Anti-MHC class I antibodies, Fc deglycosylation, and Fab production
Anti-MHC class I mouse monoclonal IgG2a antibody clones 34-1-2S, SF1.1.10, and AF6-88.5.5.3, as well as mouse IgG2a isotype control antibody clone C1.18.4, were purchased from Bio X Cell (West Lebanon, NH).
Antibodies were deglycosylated by using deGlycIT spin columns (Genovis, Lund, Sweden) containing IgGZERO (EndoS, an IgG-specific endoglycosidase acting on complex type N-glycans at the Fc glycosylation site of IgG) covalently coupled to agarose beads, according to the manufacturer’s protocol. Deglycosylation was confirmed by using reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (supplemental Figure 1A), as well as by mass spectrometry (described later) (supplemental Figure 1B).
Antibody-Fab fragments were produced from full-length IgG using the FabULOUS Fab kit (Genovis) containing cysteine protease (SpeB), according to the manufacturer’s instruction. Fab fragment size was confirmed by using nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (supplemental Figure 2).
Generation of stable H-2Kd, H-2Kb, and HLA molecules
H-2Kd–peptide complex was generated via an in vitro refolding reaction using the Luciferase peptide GFQSMYTFV (JPT Peptide Technologies GmbH, Berlin, Germany), and the H-2Kb–peptide complex was generated through UV-mediated MHC peptide exchange of Ovalbumin peptide SIINFEKL (JPT Peptide Technologies GmbH), as described previously.18 Peptide-HLA (pHLA) class I complexes pHLA-A*02:01, pHLA-A*24:02, pHLA-B*07:02, and pHLA-B*35:01 were generated through UV-mediated HLA peptide exchange of cytomegalovirus peptide NLVPMVATV (JPT Peptide Technologies GmbH), human GPR143 peptide LYSACFWWL (JPT Peptide Technologies GmbH), respiratory syncytial virus peptide NPKASLLSL (JPT Peptide Technologies GmbH), and influenza virus peptide LPFDKSTIM (JPT Peptide Technologies GmbH, Berlin, Germany), respectively. Both MHC class I–peptide complexes and all pHLA class I complexes were biotinylated as reported elsewhere.18
Surface plasmon resonance
Surface plasmon resonance (SPR) measurements were performed on an IBIS MX96 (IBIS Technologies, Enschede, The Netherlands) device as described previously by Dekkers et al.17 For measuring the affinity of the antibodies toward H-2Kd or H-2Kb, the antibodies were covalently bound on a SensEye G Easy2Spot (Ssens, Enschede, The Netherlands; 1-09-04-006). The antibodies were spotted in twofold dilutions in duplicate, ranging from 60 nM to 7.5 nM in 10 mM acetate buffer + 0.075% Tween 80, pH 5.0. The sensor was blocked with 100 mM ethanol amine, pH 8.0. Then, H-2Kd or H-2Kb was injected over the IBIS at 2 times dilution series ranging from 460 nM to 0.22 nM in phosphate-buffered saline (PBS) + 0.075% Tween 80. Regeneration after every sample was conducted with 10 mM Glycine-HCl, pH 2.0.
All C-terminally biotinylated mouse FcγRs (mFcγRs, Sino Biological, Beijing, China) were spotted by using a Continuous Flow Microspotter (Wasatch Microfluidics, Salt Lake City, UT) onto a single SensEye G-streptavidin sensor (Ssens, 1-08-04-008), allowing for binding affinity measurements of each antibody to all mFcγR simultaneously on the IBIS MX96. The biotinylated mFcγRs were spotted in threefold dilutions in duplicate, ranging from 30 nM to 1 nM for mFcγRI, mFcγRIIb, and mFcγRIV in PBS supplemented with 0.075% Tween 80, pH 7.4. Biotinylated anti-His mouse IgG1 (GenScript, Piscataway, NJ; A00613) was also spotted in this step in duplicate and threefold dilution, ranging from 10 nM to 0.3 nM. Subsequently, 100 nM His-tagged mFcγRIII was loaded onto the sensor before each antibody injection. Antibodies were then injected over the IBIS at 3 times dilution series ranging from 1000 nM to 4.1 nM in PBS + 0.075% Tween 80. Regeneration after every sample was conducted with 10 mM Glycine-HCl, pH 2.4.
Calculation of the dissociation constant (KD) was performed by equilibrium fitting to Rmax = 500 for binding of mFcγR and Rmax = 1000 for H-2Kd or H-2Kb binding. In the case of anti-His spots, association and dissociation curves of His-tagged mFcγRIII were subtracted before calculation of IgG-binding affinity using SPRINT 1.9.4.4 software (IBIS Technologies). Analysis and calculation of all binding data were performed with Scrubber software version 2 (BioLogic Software, Canberra, ACT, Australia) and Microsoft Excel (Microsoft Corporation, Redmond, WA).
Antibody Fc glycosylation analysis
Murine IgG samples (5 µg) were added to 100 µL 100 mM formic acid, followed by a 45-minute incubation at room temperature. Samples were subsequently dried by vacuum centrifugation and subjected to overnight tryptic digestion.19
Human IgG samples were extracted from TRALI donor and control plasma samples. MaxiSorp plates (Thermo Fisher Scientific, Waltham, MA) were coated overnight at 4°C with streptavidin (Thermo Fisher Scientific) 10 µg/mL in coating buffer (0.05 M carbonate-bicarbonate buffer; pH = 9.6). Plates were washed with PBS + 0.1% Tween 20 (PBS-T) and incubated for 1 hour at 37°C with biotinylated HLA molecules (HLA-A02, HLA-A24, HLA-B07, and HLA-B35) 1.55 µg/mL in PBS. Plates were washed with PBS-T and 2 times with PBS. Subsequently, plates were washed twice with 50 mM ammonium bicarbonate. Bound antibodies were eluted with 100 mM formic acid, incubated 5 minutes while shaking, and samples were transferred to a 96-well low binding plate. Samples were dried by vacuum centrifugation and subjected to overnight tryptic digestion at 60°C for ∼4 hours and trypsinized overnight with 0.01 µg/µL trypsin (Promega, Madison, WI) in 25 mM ammonium bicarbonate at 37°C. The reaction was stopped by adding formic acid to a final concentration of 2%.
IgG Fc glycosylation was assessed by analyzing tryptic (glyco)peptides with nano-LC reverse-phase–electrospray–mass spectrometry on an Impact HD quadrupole-time-of-flight mass spectrometry (Bruker Daltonics, Bremen, Germany).19 Mass spectrometry results were extracted and evaluated by using Skyline software (version 4.2.0.19107, MacCoss Lab, Department of Genome Sciences, University of Washington). The total level of glycan traits was calculated as relative intensities of IgG Fc glycopeptides obtained by integrating the first 3 isotopic peaks of double- and triple-charged glycopeptides. The level of galactosylation was calculated according to the formula (G1F + G1FN + G1FS) × 0.5 + G2F + G2FN + G2FS + G3F + G3FS. The percentage of sialylation was likewise calculated by using the formula (G1FS + G2FS + G3FS) × 0.5. For all analysis, signals from Neu5Ac- and Neu5Gc-containing species were combined. The percentage of bisecting N-acetylglucosamine (GlcNAc) was determined by summing the relative intensities of all bisected glycoforms (G0FN, G1FN, and G2FN). Similarly, the percentage of IgG fucosylation was determined by summing all fucosylated IgG Fc N-glycoforms (G0F + G1F + G0FN + G2F + G1FN + G2FN + G1FS + G2FS + G3F + G2FSαGal + GlcNAcFuc). The level of deglycosylation by EndoS treatment was determined as the relative intensity of GlcNAcFuc. In addition to the mentioned glycoforms, G0 were also detected and included in the analysis.
A large number of replicates were taken along and measurements of the precision of the methods were performed, confirming the reliability of the glycoanalytical method.
Antibody-binding and complement C1q-binding enzyme-linked immunosorbent assay
Complement C1q-binding enzyme-linked immunosorbent assay (ELISA) was adapted and modified from Dekkers et al.17 MaxiSorp plates were coated overnight at 4°C with streptavidin (Thermo Fisher Scientific) 2 µg/mL in coating buffer (0.05 M carbonate-bicarbonate buffer; pH = 9.6). Thereafter, plates were washed with PBS-T and incubated for 1 hour at 37°C with biotinylated H-2Kd or H-2Kb (387.5 ng/mL) in PBS. Plates were then washed with PBS-T and incubated for 1 hour at room temperature with indicated antibodies in a serial dilution range 1:2 starting at 3.13 µg/mL in PBS. Hereafter, the degree of antibody binding to H-2Kd or H-2Kb, and in parallel, the C1q-binding ELISA were performed.
To establish the degree of antibody binding to H-2Kd or H-2Kb, plates were washed with PBS-T and incubated for 1 hour at room temperature with goat anti-mouse κ-horseradish peroxidase (Southern Biotech, Birmingham, AL) diluted 1:1000 in PBS. Plates were washed with PBS-T, and colorimetric detection was performed by using TMB solution. Color development was stopped by using 2 M H2SO4, and the absorbance was measured by using an ELISA synergy 2 plate reader (BioTek, Winooski, VT) at 450 nm.
To establish the degree of C1q binding, after antibody incubation to H-2Kd or H-2Kb, the plates were washed with PBS-T and incubated for 1 hour at room temperature with complement serum from mouse (Sigma-Aldrich, St. Louis, MO) 1:500 in Veronal buffer with 2 mM MgCl2 and 10 mM CaCl2 and 0.1% poloxamer (Sigma-Aldrich). Hereafter, plates were incubated for 1 hour with mouse IgG2b anti-mouse C1q antibody (Invitrogen, Carlsbad, CA) 1:250 at room temperature in PBS. Plates were then washed with PBS-T and incubated for 1 hour with goat anti-mouse IgG2b–horseradish peroxidase antibody (Invitrogen) 1:2000 in PBS. Plates were washed with PBS-T, and colorimetric detection was performed by using TMB solution. Color development was stopped by using 2 M H2SO4 and the absorbance was measured by using an ELISA synergy 2 plate reader (BioTek) at 450 nm.
Statistical analysis
Statistical analysis was performed by using GraphPad Prism 8.1.2. software for Windows (GraphPad Software, San Diego, CA) with statistical significance set at P < .05. The specific statistical test applied in indicated figures and tables is listed in the accompanying legend. Standard deviations are used.
Results
34-1-2S and SF1.1.10 have similar binding affinity to BALB/c-derived H-2Kd
We investigated the biological and structural features of murine anti-MHC class I antibodies, which are known to either induce TRALI or not in BALB/c and C57BL/6 mice (Figure 1). We first tested whether the binding affinity of the TRALI-inducing 34-1-2S antibody to H-2Kd is different compared with the TRALI-resistant SF1.1.10 antibody by SPR. IgG2a 34-1-2s, IgG2a SF1.1.10, or IgG2a isotype control antibody C1.18.4. were spotted on a sensor, and a stable H-2Kd complex was subsequently flowed over the bound antibodies documenting the association-dissociation curves and the binding affinity (KD) of the antibodies toward H-2Kd. Although the association and dissociation kinetics of 34-1-2S for H-2Kd were slightly faster than those of SF1.1.10, both 34-1-2S and SF1.1.10 exhibited comparable binding affinities to H-2Kd (1.2-fold difference) (Figure 2A-B), with no binding being observed for C1.18.4 (Figure 2C).
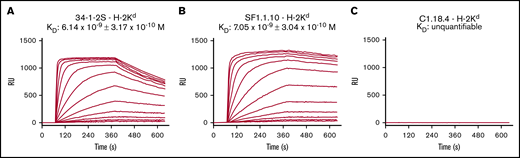
34-1-2S and SF1.1.10 both potently bind to H-2Kd. Binding affinities of IgG2a anti-MHC class I antibodies 34-1-2S (A) and SF1.1.10 (B) toward H-2Kd were determined by using SPR, and C1.18.4 (C) was used as IgG2a isotype control. Panels show binding affinity (KD) with association–dissociation curves (RU) in which antibodies were spotted in twofold dilutions in duplicate, ranging from 60 nM to 7.5 nM. A representative of n = 6 experiments is shown in each figure panel.
34-1-2S and SF1.1.10 both potently bind to H-2Kd. Binding affinities of IgG2a anti-MHC class I antibodies 34-1-2S (A) and SF1.1.10 (B) toward H-2Kd were determined by using SPR, and C1.18.4 (C) was used as IgG2a isotype control. Panels show binding affinity (KD) with association–dissociation curves (RU) in which antibodies were spotted in twofold dilutions in duplicate, ranging from 60 nM to 7.5 nM. A representative of n = 6 experiments is shown in each figure panel.
AF6-88.5.5.3 binds strongly and 34-1-2S reacts weakly to C57BL/6-derived H-2Kb
We then analyzed the binding affinity of IgG2a AF6-88.5.5.3, 34-1-2S, and C1.18.4 toward H-2Kb using SPR in the same manner performed as described earlier for H-2Kd. Only AF6-88.5.5.3 bound avidly to H-2Kb, in contrast to 34-1-2S and C1.18.4 (Figure 3). When analyzing the association–dissociation curves of these antibodies toward H-2Kb, a potent binding was evident for AF6-88.5.5.3 and a low but detectable binding was also observed for 34-1-2S. No binding was seen for C1.18.4. The cross-reactivity of 34-1-2S toward H-2Kb was further confirmed in a setting in which AF6-88.5.5.3 was spotted on the sensor and with H-2Kb being flowed over the sensor first, followed by infusion of either 34-1-2S or C1.18.4. This action showed that the H-2Kb–binding epitope for 34-1-2S is distinct from that of AF6-88.5.5.3, as 34-1-2S showed additional but weak binding (in accordance to its weak affinity) on top of the binding of AF6-88.5.5.3.
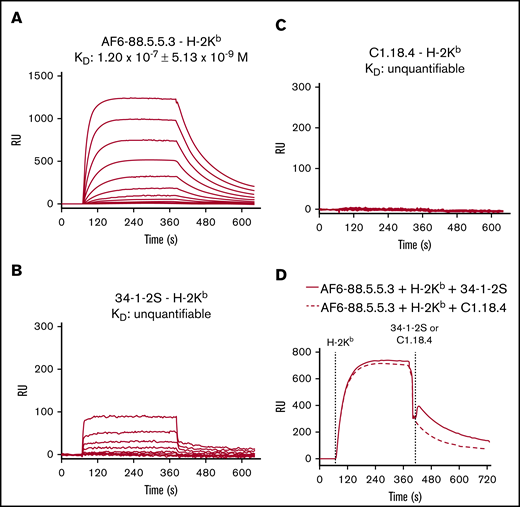
AF6-88.5.5.3 potently binds to H-2Kb, and 34-1-2S cross-reacts to H-2Kb. Binding affinities of IgG2a anti-MHC class I antibodies AF6-88.5.5.3 and 34-1-2S toward H-2Kb were determined by using SPR. C1.18.4 was used as IgG2a isotype control. (A-C) Binding affinity (KD) with association–dissociation curves (RU) is shown in which antibodies were spotted in twofold dilutions in duplicate, ranging from 60 nM to 7.5 nM. (D) Association–dissociation curves (RU) of a setup in which AF6-88.5.5.3 was spotted on the sensor, and H-2Kb was allowed to bind first, followed by infusion of either 34-1-2S or C1.18.4. A representative of n = 6 experiments is shown in each figure panel.
AF6-88.5.5.3 potently binds to H-2Kb, and 34-1-2S cross-reacts to H-2Kb. Binding affinities of IgG2a anti-MHC class I antibodies AF6-88.5.5.3 and 34-1-2S toward H-2Kb were determined by using SPR. C1.18.4 was used as IgG2a isotype control. (A-C) Binding affinity (KD) with association–dissociation curves (RU) is shown in which antibodies were spotted in twofold dilutions in duplicate, ranging from 60 nM to 7.5 nM. (D) Association–dissociation curves (RU) of a setup in which AF6-88.5.5.3 was spotted on the sensor, and H-2Kb was allowed to bind first, followed by infusion of either 34-1-2S or C1.18.4. A representative of n = 6 experiments is shown in each figure panel.
34-1-2S, SF1.1.10, and AF6-88.5.5.3 exhibit similar FcγR-binding affinity
Binding of 34-1-2S, SF1.1.10, and AF6-88.5.5.3 to murine FcγRI, FcγRIIb, FcγRIII, and FcγRIV was investigated by using SPR. The FcγRs were spotted on the sensor, and the antibodies were flowed over the sensor to measure their binding affinities. The binding differences seemed to be minor, although statistically significant (2.67-, 1.77-, 2.59-, and 1.20-fold, respectively) (Figure 4). Deglycosylated and Fab fragments of 34-1-2S, SF1.1.10, and AF6-88.5.5.3 exhibited abrogated binding to the murine FcγRs (supplemental Figures 3 and 4), compared with the untreated and full-length antibodies.
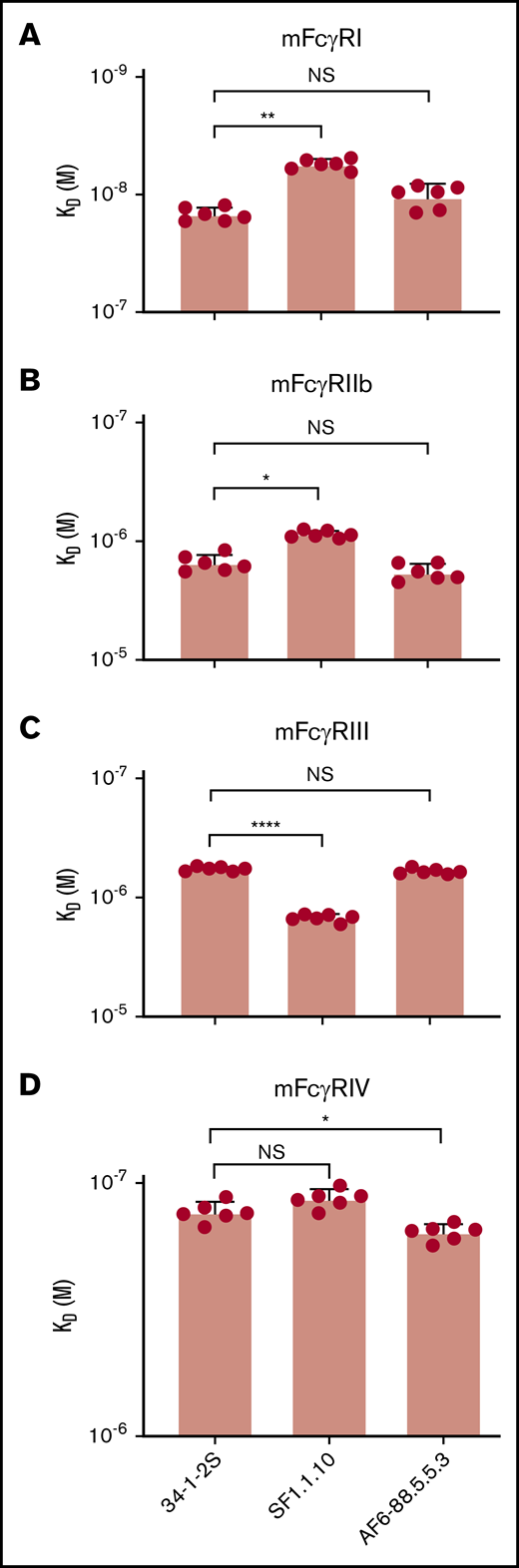
No distinct differences in FcγR-binding between 34-1-2S, SF1.1.10, and AF6-88.5.5.3. Binding affinity of IgG2a anti-MHC class I antibodies 34-1-2S, SF1.1.10, and AF6-88.5.5.3. toward murine FcγRI (A), FcγRIIb (B), FcγRIII (C), and FcγRIV (D) was determined by using SPR. Statistical analysis was performed with one-way analysis of variance with a Tukey post hoc test. *P < .05, **P < .01, ****P < .0001. A total of n = 6 experiments is depicted. NS, not significant.
No distinct differences in FcγR-binding between 34-1-2S, SF1.1.10, and AF6-88.5.5.3. Binding affinity of IgG2a anti-MHC class I antibodies 34-1-2S, SF1.1.10, and AF6-88.5.5.3. toward murine FcγRI (A), FcγRIIb (B), FcγRIII (C), and FcγRIV (D) was determined by using SPR. Statistical analysis was performed with one-way analysis of variance with a Tukey post hoc test. *P < .05, **P < .01, ****P < .0001. A total of n = 6 experiments is depicted. NS, not significant.
Fc glycosylation composition of 34-1-2S does not reveal a distinct pattern compared with SF1.1.10 and AF6-88.5.5.3
We assessed the N-linked glycan composition at site Asn297 of the Fc portion of 34-1-2S, SF1.1.10, and AF6-88.5.5.3. More specifically, the degree of fucosylation, galactosylation, sialylation, and bisecting GlcNAc was evaluated as these glycans may be variably present (Figure 5A; supplemental Figure 5). Overall, no particularly skewed pattern of 34-1-2S-Fc glycosylation was observed compared with SF1.1.10 and AF6-88.5.5.3; a high degree of Fc fucosylation was present for all antibodies (Table 1). Notably, however, the degree of 34-1-2S Fc-galactosylation was slightly lower compared with the other antibodies.
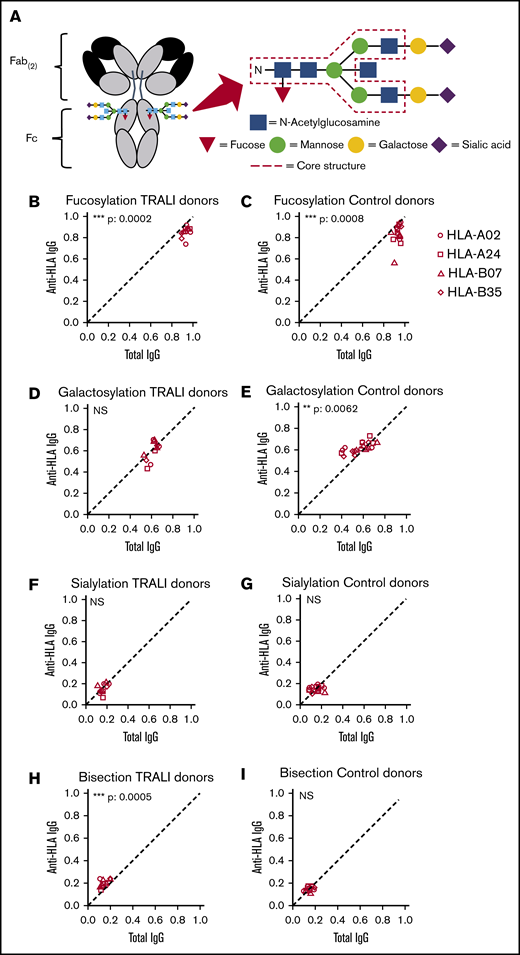
Anti-HLA Fc Asparagine (N)-297-linked glycan structures are similar in TRALI donors compared with control donors. (A) The conserved IgG-Fc glycan core structure consists of a bi-antennary glycan core of GlcNAc and mannose. In addition, glycans can be variably present such as fucose, galactose, sialic acid, and bisecting GlcNAc. Relative expression levels of major IgG-Fc Asn297 glycoforms for both total anti-HLA IgG1 (x-axis) and antigen-specific HLA IgG1 (y-axis) for 11 TRALI donor samples (B,D,F,H) and 20 control donor samples (C,E,G,I). HLA specificities include HLA-A02 HLA-A24, HLA-B07, and HLA-B35. Samples were analyzed for Fc fucosylation (B-C), galactosylation (D-E), sialylation (F-G), and bisection (H-I). The statistical outcome between 2-tailed paired Student t test analysis of total IgG1 vs specific antibodies is listed in each panel. The diagonal, dotted line represents the equal ratio between total IgG1 and the specific antibody. **P < .01, ***P < .001.
Anti-HLA Fc Asparagine (N)-297-linked glycan structures are similar in TRALI donors compared with control donors. (A) The conserved IgG-Fc glycan core structure consists of a bi-antennary glycan core of GlcNAc and mannose. In addition, glycans can be variably present such as fucose, galactose, sialic acid, and bisecting GlcNAc. Relative expression levels of major IgG-Fc Asn297 glycoforms for both total anti-HLA IgG1 (x-axis) and antigen-specific HLA IgG1 (y-axis) for 11 TRALI donor samples (B,D,F,H) and 20 control donor samples (C,E,G,I). HLA specificities include HLA-A02 HLA-A24, HLA-B07, and HLA-B35. Samples were analyzed for Fc fucosylation (B-C), galactosylation (D-E), sialylation (F-G), and bisection (H-I). The statistical outcome between 2-tailed paired Student t test analysis of total IgG1 vs specific antibodies is listed in each panel. The diagonal, dotted line represents the equal ratio between total IgG1 and the specific antibody. **P < .01, ***P < .001.
Fc glycosylation (%) of 34-1-2S, SF1.1.10, and AF6-88.5.5.3
| Fc glycosylation | 34-1-2S | SF1.1.10 | AF6-88.5.5.3 |
|---|---|---|---|
| Fucosylation | 99.87 | 99.86 | 99.76 |
| Galactosylation | 11.67 | 20.69 | 17.47 |
| Sialylation | 0.27 | 2.20 | 0.57 |
| Bisecting GlcNAc | 1.28 | 1.09 | 1.17 |
| Fc glycosylation | 34-1-2S | SF1.1.10 | AF6-88.5.5.3 |
|---|---|---|---|
| Fucosylation | 99.87 | 99.86 | 99.76 |
| Galactosylation | 11.67 | 20.69 | 17.47 |
| Sialylation | 0.27 | 2.20 | 0.57 |
| Bisecting GlcNAc | 1.28 | 1.09 | 1.17 |
TRALI-causing donor anti-HLA antibodies exhibit comparable Fc glycosylation profiles as control donor anti-HLA antibodies
We also examined the Fc glycosylation profiles of donor anti-HLA antibodies, which were involved in triggering a TRALI reaction (n = 11), compared with control donor anti-HLA antibodies (n = 20). When comparing antigen-specific IgG1 (HLA-A02, HLA-A24, HLA-B07, and HLA-B35) vs total IgG1, we found that anti-HLA antibodies from both TRALI and control donors exhibited a slight but significant decrease in Fc fucosylation. In addition, a relatively decreased level of galactosylation was observed for total IgG of control donors and a relatively increased anti-HLA Fc bisection for TRALI donors (Figure 5B-I; Table 2). When directly comparing the Fc glycosylation profiles of TRALI-inducing anti-HLA antibodies vs control anti-HLA antibodies, no significant differences were observed for fucosylation, galactosylation, and sialylation; only an increased bisection for TRALI-inducing antibodies was measured (****P < 0.0001).
Fc glycosylation (relative expression levels) of total IgG1 vs specific IgG1 of TRALI donor anti-HLA antibodies vs control donor anti-HLA antibodies
| Fc glycosylation | Total IgG1 | Specific IgG1 | Total IgG1 vs Specific IgG1, P |
|---|---|---|---|
| TRALI donor anti-HLA antibodies | |||
| Fucosylation | 0.93 ± 0.03 | 0.85 ± 0.05 | P = .0002, *** |
| Galactosylation | 0.61 ± 0.05 | 0.60 ± 0.09 | NS |
| Sialylation | 0.17 ± 0.03 | 0.16 ± 0.05 | NS |
| Bisecting GlcNAc | 0.15 ± 0.03 | 0.20 ± 0.03 | P = .0005, *** |
| Control donor anti-HLA antibodies | |||
| Fucosylation | 0.94 ± 0.03 | 0.86 ± 0.09 | P = .0008, *** |
| Galactosylation | 0.57 ± 0.10 | 0.62 ± 0.05 | P = .0062, ** |
| Sialylation | 0.15 ± 0.05 | 0.15 ± 0.02 | NS |
| Bisecting GlcNAc | 0.15 ± 0.03 | 0.15 ± 0.02 | NS |
| Fc glycosylation | Total IgG1 | Specific IgG1 | Total IgG1 vs Specific IgG1, P |
|---|---|---|---|
| TRALI donor anti-HLA antibodies | |||
| Fucosylation | 0.93 ± 0.03 | 0.85 ± 0.05 | P = .0002, *** |
| Galactosylation | 0.61 ± 0.05 | 0.60 ± 0.09 | NS |
| Sialylation | 0.17 ± 0.03 | 0.16 ± 0.05 | NS |
| Bisecting GlcNAc | 0.15 ± 0.03 | 0.20 ± 0.03 | P = .0005, *** |
| Control donor anti-HLA antibodies | |||
| Fucosylation | 0.94 ± 0.03 | 0.86 ± 0.09 | P = .0008, *** |
| Galactosylation | 0.57 ± 0.10 | 0.62 ± 0.05 | P = .0062, ** |
| Sialylation | 0.15 ± 0.05 | 0.15 ± 0.02 | NS |
| Bisecting GlcNAc | 0.15 ± 0.03 | 0.15 ± 0.02 | NS |
Statistical analysis: 2-tailed paired Student t test. **P < .01, ***P < .001.
H-2Kd–bound 34-1-2S shows increased complement activation, which is fully Fc mediated but not significantly dependent on Fc glycosylation
To investigate if TRALI-inducing antibodies have an increased ability to activate complement, we used a previously established antibody-based C1q-binding ELISA.17 In this ELISA setup, we first confirmed that both 34-1-2S and SF1.1.10 antibodies exhibited equal degrees of binding to H-2Kd (Figure 6A). Despite equal antigen binding, 34-1-2S demonstrated increased complement activation compared with SF1.1.10. Because Fc glycans may also influence complement activation, antibodies were deglycosylated (supplemental Figure 1) and tested for their ability to activate complement. Deglycosylation of 34-1-2S did not significantly decrease complement activation (despite a decreased trend), whereas deglycosylation of SF1.1.10 did significantly decrease the activation of complement compared with untreated SF1.1.10. Deglycosylated 34-1-2S was still able to bind more C1q than deglycosylated SF1.1.10. Importantly, the deglycosylation did not affect binding of these antibodies to H-2Kd, as expected. Furthermore, antibody-mediated complement activation was fully Fc mediated, as antibody-Fab fragments (supplemental Figure 2) were unable to activate complement with a similar degree of binding to H-2Kd (apart from a somewhat higher degree of SF1.1.10–Fab binding). As a negative control, heat inactivation was applied to the complement serum, which also fully abrogated C1q binding.
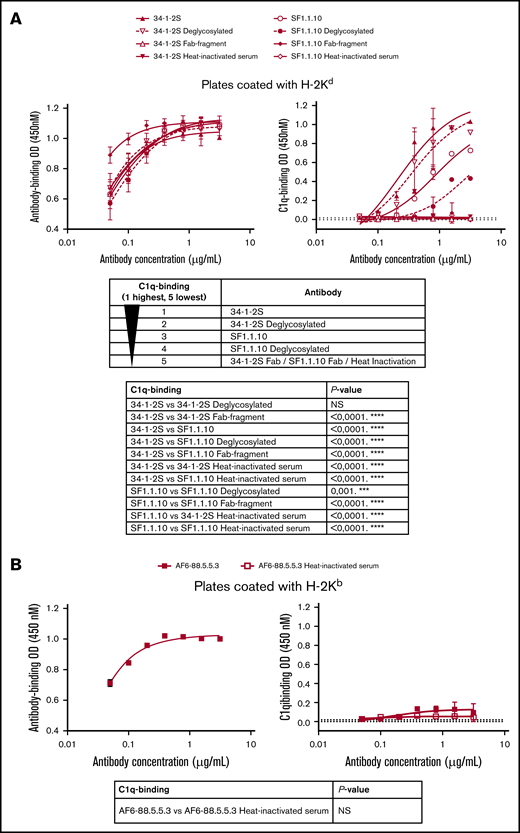
34-1-2S demonstrates increased complement activation compared with SF1.1.10 and AF6-88.5.5.3 that is Fc dependent and not significantly dependent on Fc glycosylation. Antibody- and C1q-binding ELISAs based on 34-1-2S and SF1.1.10 (full-length IgG, deglycosylated IgG, Fab fragments, and with a serum heat-inactivation control) binding to H-2Kd (A, left and right panel, respectively). Antibody- and C1q-binding ELISAs based on AF6-88.5.5.3 (full-length IgG and with serum heat-inactivation control) binding to H-2Kb (B, left and right panel, respectively). Statistical analysis for C1q binding is performed with two-way analysis of variance with a Tukey post hoc test (A) and an ordinary 2-way analysis of variance (B). A representative ELISA of n = 5 experiments is shown in each figure panel. ***P < .001, ****P < .0001.
34-1-2S demonstrates increased complement activation compared with SF1.1.10 and AF6-88.5.5.3 that is Fc dependent and not significantly dependent on Fc glycosylation. Antibody- and C1q-binding ELISAs based on 34-1-2S and SF1.1.10 (full-length IgG, deglycosylated IgG, Fab fragments, and with a serum heat-inactivation control) binding to H-2Kd (A, left and right panel, respectively). Antibody- and C1q-binding ELISAs based on AF6-88.5.5.3 (full-length IgG and with serum heat-inactivation control) binding to H-2Kb (B, left and right panel, respectively). Statistical analysis for C1q binding is performed with two-way analysis of variance with a Tukey post hoc test (A) and an ordinary 2-way analysis of variance (B). A representative ELISA of n = 5 experiments is shown in each figure panel. ***P < .001, ****P < .0001.
H-2Kb–bound AF6-88.5.5.3 is incapable of complement activation
Similarly, using the antibody-dependent C1q-binding ELISA, we tested the complement activation by H-2Kb–bound AF6-88.5.5.3. In this ELISA setup, we confirmed that AF6-88.5.5.3 was able to strongly bind to H-2Kb (Figure 6B). H-2Kb–bound AF6-88.5.5.3 was incapable of binding C1q, as this occurred similarly as the negative control in which heat-inactivated complement serum was added to H-2Kb–bound AF6-88.5.5.3.
Discussion
TRALI remains one of the leading causes of transfusion-related mortality, and preventive or therapeutic strategies are not available.1,2 The pathogenesis remains a challenge to decipher; however, murine models of antibody-mediated TRALI have provided significant mechanistic insights into the disease pathology. Curiously, only certain anti-MHC class I antibodies cause TRALI in mice, whereas other anti-MHC class I antibodies (also IgG2a) are unable to induce TRALI.7 In BALB/c (H-2Kd) TRALI mouse models, the anti-MHC class I antibody 34-1-2S (IgG2a, anti–H-2Kd) is classically used to induce severe TRALI, whereas another well-known anti-MHC class I antibody, SF1.1.10 (also IgG2a and H-2Kd specific), is unable to trigger murine TRALI. For the TRALI model in C57BL/6 mice (H-2Kb), only a combination of anti-MHC class I antibodies AF6-88.5.5.3 (IgG2a, anti–H-2Kb) with a higher dose of 34-1-2S causes TRALI.4 The biological reasons for this action are unknown.
The current article focused on the biological and structural antibody characteristics that could provide mechanistic explanations for the requirements of antibody-mediated onset of TRALI in mice. We found that 34-1-2S or SF1.1.10 both potently bound to H-2Kd. Similarly, AF6-88.5.5.3, which on its own does not cause TRALI in mice, bound potently to H-2Kb while 34-1-2S bound weakly and cross-reacted with H-2Kb. In addition, the H-2Kb–binding epitope for 34-1-2S was shown to be distinct from that of AF6-88.5.5.3, as both antibodies could bind H-2Kb simultaneously. The observed cross-reactivity of 34-1-2S to H-2Kb was in line with a previous study of Ozato et al.20 Collectively, this indicates that antigen-binding affinity is not driving the antibody-mediated TRALI induction in mice.
Because FcγRs were previously suggested to be involved in murine TRALI,5 although their involvement was deemed less important by another study,7 we investigated the ability of the murine anti-MHC class I antibodies to bind to murine FcγRs using SPR. In general (despite some minor variations), we found no substantial differences between these antibodies in binding to FcγRs that could explain the TRALI-inducing capability. FcγRs may still be involved in the pathogenesis of TRALI, and this possibility should be further investigated in subsequent studies. These studies should also take into account that wild-type mice lack the FcγRIIa that may potentially have an important role in human TRALI, as was shown for platelet activation by circulating immune complexes in a setting of systemic shock.21
Because IgG-Fc glycosylation is also known to significantly influence interactions with FcγRs, we also analyzed the Fc glycosylation patterns of the anti-MHC class I antibodies. Using mass spectrometry, we evaluated the composition of Fc glycans, which can differ depending on the setting.22,23 For instance, a low Fc-core fucosylation has been observed for anti-human platelet antigen-1a antibodies in fetal and neonatal alloimmune thrombocytopenia, in contrast to anti-HLA antibodies in refractory thrombocytopenia; this low fucose was subsequently shown to enhance the Fc-binding affinity to FcγRIIIa/b, thereby increasing antibody-mediated platelet phagocytosis.24 This IgG-glycosylation pattern also correlated with clinical disease severity in fetal and neonatal alloimmune thrombocytopenia,24,25 but other factors were also found to be important such as Fc galactosylation and antibody titer.25 Another similar example for low core Fc fucosylation was found for IgG antibodies to dengue that enhanced binding to FcγRIIIa and also determined disease severity.26 In this study, however, we found no deviant IgG-glycosylation pattern for 34-1-2S, and the high degree of Fc fucosylation in all antibodies further supports that no meaningful biological differences for binding to mouse FcγRIV were observed.27,28 Also upon analysis of the Fc glycosylation profiles of TRALI-inducing donor anti-HLA antibodies compared with control donor anti-HLA antibodies, overall no substantial differences were observed. Notably, when comparing total IgG vs specific anti-HLA IgG, total IgG galactosylation in control donors was relatively decreased, which may be related to postmenopausal changes (data not shown). Also, in TRALI donors, anti-HLA Fc bisection was found to be increased compared with total IgG. To the best of our knowledge, no functional implications have been revealed for increased IgG Fc glycan bisection. Remarkably, however, a slightly lowered Fc fucosylation was observed for both TRALI and control donor anti-HLA antibodies. This finding indicates that anti-HLA antibodies can display a low Fc fucose; however, it seems that this action is not necessarily required for TRALI induction. The finding that Fc fucosylation is not a critical factor for TRALI induction is also supported by the mouse IgG-Fc glycosylation data, which showed high levels of Fc fucose in all tested antibodies, regardless of their capability to induce TRALI. Anti-HLA antibodies also play a role in kidney transplantation, but their glycosylation has hitherto not been evaluated as a risk factor. Our data indicate that anti-HLA antibodies may show diverse fucosylation levels, and a broader evaluation of anti-HLA Fc glycosylation as a pathology and risk factor is warranted beyond the field of transfusion.
Because complement involvement has not been thoroughly investigated in TRALI,16 and its involvement may be plausible,7 we tested the ability of anti-MHC class I antibodies to activate complement in vitro. Antibodies are the main drivers of the classical complement-activating pathway, which can be activated by binding of C1q to IgG.16 We therefore investigated antibody–C1q binding by using an established C1q-binding ELISA,17 based on antibody binding to H-2Kdor H-2Kb. We found H-2Kd–bound 34-1-2S to be more potent in complement activation compared with H-2Kd–bound SF1.1.10. This effect was fully Fc dependent, as antibody Fab fragments no longer bound C1q (despite a seemingly higher binding of SF1.1.10-Fab to H-2Kd). This finding is in line with the landmark article of Diebolder et al,29 who described that antibody-mediated complement activation occurs via specific noncovalent Fc interactions resulting in formation of ordered antibody hexamers upon antigen binding. H-2Kd–bound 34-1-2S may therefore be able to more efficiently form hexamers than H-2Kd–bound SF1.1.10. In addition, Fc glycans may affect complement activation, as elevated human antibody Fc galactosylation and sialylation were shown to significantly increase C1q binding.17 We therefore tested deglycosylated anti-MHC class I antibodies. We found that for 34-1-2S, this did not significantly decrease C1q binding in contrast to deglycosylated SF1.1.10, with H-2Kd–bound 34-1-2S and deglycosylated 34-1-2S still binding more C1q than H-2Kd–bound SF1.1.10; and deglycosylated SF1.1.10. H-2Kb–bound AF6-88.5.5.3 was found to be incapable of inducing complement, in line with its inability to induce TRALI, despite its potent binding to H-2Kb. Thus, 34-1-2S may perhaps be additionally required to increase complement levels for the onset of TRALI in C57BL/6 mice. In line with this, it was recently shown that combinations of anti-HLA antibodies30 or anti-CD20 antibodies together with anti-CD37 antibodies31 could synergistically activate complement. The requirement for a higher dose of 34-1-2S in C57BL/6 mice may be needed to strengthen the relatively weak cross-reactive binding to H-2Kb. The degree of 34-1-2S Fc galactosylation was slightly lower compared with the other antibodies, but this difference may not have been a biologically substantial difference, as for human antibodies a lower Fc galactosylation was shown to correspond to less C1q-binding.17 Future studies, however, should assess the role of Fc galactosylation in C1q binding for murine antibodies.
Strait et al7 previously injected BALB/c mice intravenously with 34-1-2S, SF1.1.10, or control antibody. After 3 minutes, mononuclear cells were isolated from blood and analyzed for C3 binding by using flow cytometry. In line with our data, it was found that 34-1-2S infusion resulted in increased (∼2 times) mononuclear cell binding of C3 compared with infusions with SF1.1.10. These data will need to be validated and further explored in future animal and human studies. For the human TRALI studies, it should be taken into account that complement may also be activated as a first hit in the recipient, and therefore pretransfusion and posttransfusion complement analyses will also be important.
In conclusion, we found that the pathogenic nature of murine TRALI-causing antibodies does not seem to be specifically related to the MHC-class I binding affinity, binding to FcγRs or to the Fc glycosylation composition. The TRALI antibody pathogenicity seems to be associated with an increased ability to activate complement, which is fully Fc dependent and not significantly dependent upon Fc glycosylation. This does not necessarily indicate that increased complement activation explains all of the pathogenicity but that it may be one of the essential TRALI antibody characteristics. Further research should focus on complement involvement in patients with TRALI, while also taking non-antibody triggers into consideration.
Requests for data sharing may be submitted to the corresponding author (Rick Kapur; e-mail: r.kapur@sanquin.nl).
Acknowledgments
The authors thank Agnes Hipgrave Ederveen for expert technical support.
This work was supported by Sanquin (grant PPOC-18-08).
Authorship
Contribution: E.A.N.Z.v.d.L., S.v.d.V., A.E.H.B., T.L.J.v.O., J.Y.M., G.B., and D.M.G. performed experiments and collected data; M.D.L. performed mouse antibody glycosylation analysis; C.A.M.K. and J.N. performed human antibody glycosylation analysis; R.K. conceived and supervised the study, and wrote the paper; and all authors (including J.W.S., L.P., W.J.E.v.E., M.W., C.E.v.d.S., G.V., and R.K.) analyzed and interpreted data and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rick Kapur, Department of Experimental Immunohematology, Sanquin Research, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: r.kapur@sanquin.nl.

