

Key Points
Missense mutations in the FIX signal peptide and propeptide cause FIX deficiency by various mechanisms.
Oral administration of vitamin K may alleviate the severity of hemophilia B with certain missense mutations in FIX.

Abstract
Many mutations in the signal peptide and propeptide of factor IX (FIX) cause hemophilia B. A FIX variants database reports 28 unique missense mutations in these regions that lead to FIX deficiency, but the underlying mechanism is known only for the mutations on R43 that interfere with propeptide cleavage. It remains unclear how other mutations result in FIX deficiency and why patients carrying the same mutation have different bleeding tendencies. Here, we modify a cell-based reporter assay to characterize the missense mutations in the signal peptide and propeptide of FIX. The results show that the level of secreted conformation-specific reporter (SCSR), which has a functional γ-carboxyglutamate (Gla) domain of FIX, decreases significantly in most mutations. The decreased SCSR level is consistent with FIX deficiency in hemophilia B patients. Moreover, we find that the decrease in the SCSR level is caused by several distinct mechanisms, including interfering with cotranslational translocation into the endoplasmic reticulum, protein secretion, γ-carboxylation of the Gla domain, and cleavage of the signal peptide or propeptide. Importantly, our results also show that the SCSR levels of most signal peptide and propeptide mutations increase with vitamin K concentration, suggesting that the heterogeneity of bleeding tendencies may be related to vitamin K levels in the body. Thus, oral administration of vitamin K may alleviate the severity of bleeding tendencies in patients with missense mutations in the FIX signal peptide and propeptide regions.
Introduction
Hemophilia B is an X-linked recessive bleeding disorder with a prevalence rate of ∼1 in 30 000 live male births.1 This inherited disorder is caused by deficiency of coagulation factor IX (FIX) in patients, whose bleeding tendencies correlate with their plasma level of FIX activity (FIX:C). With the normal level of FIX:C at 1 IU/mL in plasma, patients having <1% of normal FIX:C (<0.01 IU/mL) are defined as severe cases, those with 1% to 5% of normal (0.01-0.05 IU/mL) are considered moderate cases, and those with 5% to 40% of normal (0.05-0.40 IU/mL) are classified as mild cases.2
A total of 1113 unique mutations in the FIX gene had been reported in hemophilia B patients.3 These mutations are distributed over exons, introns, or untranslated regions of the FIX gene, and 923 mutations occur in the coding region of FIX domains,3 which contain an N terminus signal peptide and propeptide, followed by the mature protein that is subdivided into the γ-carboxyglutamate (Gla) domain, 2 EGF domains, linkers, and the protease domain.3-5 Mutations in the signal peptide and propeptide account for 6.5% of the total mutations in the coding region of FIX.3 To date, 28 unique missense mutations have been recorded in the signal peptide and propeptide in a FIX variants database.3 Among these, the R43W and R43Q mutations render the propeptide uncleaved, resulting in deficient generation of FIX.6-9 However, it remains unclear how the other mutations affect the activity of FIX and why, in some cases, patients carrying the same mutation exhibit different bleeding tendencies that vary from mild to severe.3,5,10
The signal peptide and propeptide are required for the maturation of FIX protein.5 The signal peptide directs the cotranslational translocation of FIX into the endoplasmic reticulum (ER) lumen of liver cells; it is subsequently removed by the signal peptidase at the C28 position. The propeptide is required for γ-carboxylation of FIX, a posttranslational modification that is essential for its membrane-associated activity.11,12 The propeptide provides the primary binding site between FIX and vitamin K–dependent γ-glutamyl carboxylase (GGCX),13 which converts glutamate residues to Gla in FIX’s Gla domain. The presence of the propeptide also increases the enzymatic activity of GGCX.13,14 After γ-carboxylation of the Gla domain, the propeptide is cleaved off to generate the mature FIX protein.6
The γ-carboxylated Gla domain anchors FIX to broken cell membranes in case of injury.11,15,16 The negative charge of the broken membrane triggers the binding of Gla domain to membrane in a calcium-dependent interaction.11,12,17,18 Gla residues have a high affinity for calcium ions. In turn, the calcium binding induces a structural transition in the Gla domain, which is converted from a largely unfolded and nonfunctional19 region to a tightly folded domain that is capable of membrane binding.8,9,20 The N terminus of the folded Gla domain contains a ω-loop,8,9 which forms an exposed hydrophobic patch that participates in membrane binding.9,21,22 Conformational-specific antibody recognizing the ω-loop23 can detect whether FIX has a functional Gla domain that is fully γ-carboxylated and calcium stabilized.15,24
Here, we modify a well-established cell-based assay to characterize 28 missense mutations in the signal peptide and propeptide of FIX. We find that FIX deficiency of these missense mutations is caused by various mechanisms. In addition, our results indicate that the heterogeneity of bleeding tendencies in hemophilia B patients with the same mutation may be caused by different levels of vitamin K in the body, suggesting that oral administration of vitamin K may be a strategy to alleviate the severity of bleeding in patients with certain missense mutations in FIX signal peptide and propeptide.
Materials and methods
Research subjects and FIX mutations nomenclature
The missense mutations in the signal peptide and propeptide regions were analyzed based on the online FIX variants database (http://www.factorix.org/index.php#).3 Detailed information about patients was collected (supplemental File 1), and mutations in the FIX protein were described following the nomenclature system of the Human Genome Variation Society (http://www.hgvs.org/mutnomen). The sequence NP_000124.1 of FIX protein was used as a reference for amino acid numbers; mature FIX starts at residue 47.
Plasmid construction
A chimera reporter, termed “FIX-PC,” was constructed with the N-terminal domains of FIX (signal peptide, propeptide, and Gla domain) fused to the C-terminal domains of protein C (supplemental Figure 1A), which is similar to a previous reporter.15,25 This reporter protein allows us to detect the calcium-stabilized conformation of FIX’s Gla domain by using a monoclonal antibody. This reporter gene was subcloned into the EF1α promoter multiclone site of the pBud CE4.1 vector. To evaluate the transfection efficiency, Metridia luciferase complementary DNA was subcloned into the same vector at the CMV promoter multicloning site (supplemental Figure 1B). The mutants were created using a ligation-free method, as previously described.26
Detection of the secreted FIX-PC reporter in medium
HEK293T cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (both from Gibco). The cells were plated onto a 24-well plate to reach ∼50% to 60% confluence on the following day, and then cells were transfected with different constructs using Lipofectamine 3000 (Thermo Fisher Scientific). After 4 to 6 hours of transfection, the medium was replaced by fresh complete medium supplemented with vitamin K, the cells were incubated for an additional 36 to 48 hours, and the medium was collected. Calcium ion was added to the medium to 5 mM final concentration. The secreted conformation-specific reporter (SCSR) of FIX-PC was detected by an antibody (GMA001; Green Mountain Antibodies), as previously described.15
To detect the secreted total reporter (STR) of FIX-PC, each well of a 96-well enzyme-linked immunosorbent assay plate was coated with 100 μL of mouse anti–human protein C monoclonal antibody (2 μg/mL; GMA-067; Green Mountain Antibodies) overnight at 4°C. After washing with TBS with Tween 20 buffer (20 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 0.1% Tween 20) 5 times, the plate was blocked using bovine serum albumin. The samples and FIX-PC protein standards were added and incubated for 2 hours at room temperature. After the unbound proteins were washed off using TBS with Tween 20 buffer 5 times, 100 μL of sheep anti–human protein C immunoglobulin G conjugated to horseradish peroxidase (HRP) (HRP1598-1R4; Affinity Biologicals) was added to each well and incubated for an additional hour at room temperature. After unbound antibody was washed off, ABTS substrate was used for color development, and absorbance was measured at 405 nm by a plate reader.
To eliminate the transfection efficiency difference among constructs, SCSR and STR levels of different samples were divided by their Metridia luciferase, which was measured as previously described27 (supplemental Figure 1C). These corrected SCSR and STR levels were further normalized to those of FIX-PC with wild-type signal peptide and propeptide sequences.
Normalization of the SCSR/STR ratio of FIX-PC
To analyze the relationship between SCSR and STR of FIX-PC, we calculated the ratio of SCSR/STR (RC/T). RC/T of mutants was normalized to that of wild-type FIX-PC. All error propagations were calculated accordingly.
Analysis of the expression, maturation, and γ-carboxylation of FIX-PC reporter by western blotting
To analyze the expression, maturation, and γ-carboxylation of the FIX-PC reporter, the constructs were transiently expressed in HEK293T cells in a 6-well plate. Transfection and cell growth were performed as described above. The cells were washed once with ice-cold phosphate-buffered saline and lysed with 120 µL of lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail). The cell lysate was subjected to reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and western blot was conducted with sheep anti–human protein C immunoglobulin G conjugated to HRP, anti-Gla (3570; BioMedica Diagnostics), and anti–β-actin (Santa Cruz Biotechnology) antibodies. Corresponding HRP-conjugated secondary antibodies were used to detect the primary antibody, and the chemiluminescent signal was detected using an ECL kit (WBKLS0100; MilliporeSigma).
Analysis of the relative expression level and the relative γ-carboxylation efficiency of intracellular FIX-PC reporter
To determine the relative expression level of the intracellular FIX-PC reporter, the band intensity from western blot was analyzed using ImageJ (National Institutes of Health). The expression ratio of the reporter to β-actin of different mutants was normalized to that of wild-type FIX-PC to give the relative expression level.
To analyze the relative γ-carboxylation efficiency of the intracellular FIX-PC reporter, the γ-carboxylated reporter/total reporter ratio was calculated. The ratio of the mutations was normalized to that of wild-type FIX-PC to give the relative γ-carboxylation efficiency.
Signal peptide analysis
The online SignalP server (http://www.cbs.dtu.dk/services/SignalP/)28 was used to predict the probability of signal peptide in mutations.
Results
Survey of missense mutations in signal peptide and propeptide of FIX in hemophilia B patients
Based on the FIX variants database, the occurrence and frequency of missense mutations in the signal peptide and propeptide regions were investigated first. Twenty-eight unique missense mutations were reported in 2 regions, accounting for 226 cases of hemophilia B. These mutations are distributed on 9 residues in the signal peptide and 8 residues in the propeptide (Figure 1; supplemental Figure 2). In particular, 166 patients carry mutations on R43 in the propeptide, and there are 3 types of mutations (R43W/Q/L). Residue C28 at the signal peptide cleavage site is another mutation hotspot, and 4 types of mutations (C28R/G/Y/W) were found in 16 patients. Multiple mutations have also been reported for other residues, including V30 (I or L), A37 (V, T, or D), and R46 (K, T, or S), all of which are located in the propeptide region. The remaining 12 residues only have a single type of mutation that is distributed sporadically in the signal peptide and propeptide regions.

Survey of hemophilia B patients with the missense mutations in the signal peptide and propeptide. Twenty-eight unique missense mutations (226 cases) in the signal peptide and propeptide were found in the FIX variants database. Mutations on residues 1 to 28 are located in the signal peptide region, and mutations on residues 29 to 46 reside in the propeptide region.
Survey of hemophilia B patients with the missense mutations in the signal peptide and propeptide. Twenty-eight unique missense mutations (226 cases) in the signal peptide and propeptide were found in the FIX variants database. Mutations on residues 1 to 28 are located in the signal peptide region, and mutations on residues 29 to 46 reside in the propeptide region.
Characterization of the secreted reporter of the missense mutations in the signal peptide and propeptide
Mutations in the signal peptide and propeptide may allow some functional FIX to be secreted, because they are not located in the region of the mature protein. One indicator of functional FIX is its fully γ-carboxylated and properly folded Gla domain. Thus, we evaluated whether these mutations have a functional Gla domain by measuring their SCSR level.15,25 We found that the SCSR level was significantly decreased in most mutations (Figure 2A), which is consistent with FIX deficiency in hemophilia B patients.
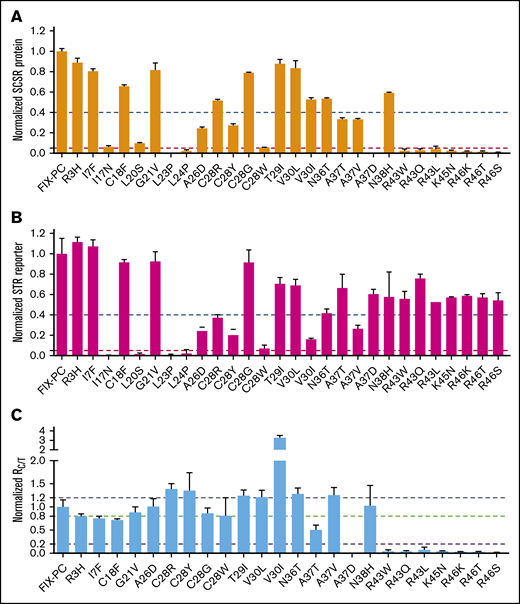
Characterization of the secreted reporter with the missense mutations in the signal peptide and propeptide. SCSR protein (A) and STR protein (B) were normalized to Metridia luciferase and expressed relative to wild-type FIX-PC. The error bars indicate the standard deviation from 3 biological replicates. The dashed blue and red lines show 40% and 5% level of wild-type FIX-PC reporter, respectively. (C) Normalized RC/T. The ratio was renormalized by RC/T of wild-type FIX-PC, which is defined as “1”. All error propagations were calculated accordingly. The black, green, and violet dashed lines show 1.2-fold, 0.8-fold, and 0.2-fold wild-type RC/T, respectively.
Characterization of the secreted reporter with the missense mutations in the signal peptide and propeptide. SCSR protein (A) and STR protein (B) were normalized to Metridia luciferase and expressed relative to wild-type FIX-PC. The error bars indicate the standard deviation from 3 biological replicates. The dashed blue and red lines show 40% and 5% level of wild-type FIX-PC reporter, respectively. (C) Normalized RC/T. The ratio was renormalized by RC/T of wild-type FIX-PC, which is defined as “1”. All error propagations were calculated accordingly. The black, green, and violet dashed lines show 1.2-fold, 0.8-fold, and 0.2-fold wild-type RC/T, respectively.
FIX deficiency in patients may be caused by the absence of, or decrease in, secreted total FIX protein.29 Thus, we measured the STR levels of the mutants and then compared RC/T. Our results show that mutations in the signal peptide region decrease SCSR protein and STR protein proportionally, with RC/T close to 1 (Figure 2B-C). Similarly, the SCSR and STR levels of several propeptide mutations, including T29I, V30L, N36T, A37V, and N38H, are proportionally expressed (Figure 2B-C). To our surprise, several mutations in the propeptide region changed the SCSR and STR levels to different extents (Figure 2B-C). V30I mutation shows increased RC/T, indicating relatively more secretion of functional protein, whereas the remaining mutations in the propeptide region show decreased RC/T, indicating relatively less secretion of functional protein. In summary, these results suggest that mutations in the signal peptide region impair FIX function, primarily by affecting the production of FIX, whereas mutations in the propeptide region may use multiple mechanisms to cause FIX deficiency.
Mutations in the hydrophobic region of signal peptide hinder cotranslational translocation of the FIX-PC reporter into the ER
The decreased SCSR level may originate from the decreased protein expression. To test that possibility, we analyzed the intracellular reporter protein expression level of the mutations by western blot. Most mutations did not show any change in this level, indicating that the decrease in their SCSR is not caused by the change in protein expression. However, certain mutations in the signal peptide (I17N, L20S, L23P, L24P, and C28Y/R/W) and the propeptide (V30I) regions reduced the reporter protein expression level (supplemental Figure 3), which is consistent with the decrease in the SCSR level (Figure 2A). Among them, 4 mutations in the signal peptide region show a lower molecular weight (MW) band compared with the other constructs (supplemental Figure 3A), which indicates that these 4 mutations have a distinct mechanism to decrease the SCSR level.
Residues of I17, L20, L23, and L24 belong to a hydrophobic region of the signal peptide (Figure 3A), which involves cotranslational translocation of FIX into the ER lumen, where MW of FIX is increased because of glycosylation. By predicting the signal peptide probability with SignalP server,28 these 4 mutations were also found to decrease the probability of signal peptide (Figure 3B). Thus, we assume that the lower MW band of these mutations may be caused by the dysfunction of signal peptide, which leads to the eradication of cotranslational translocation of the reporter into the ER lumen.
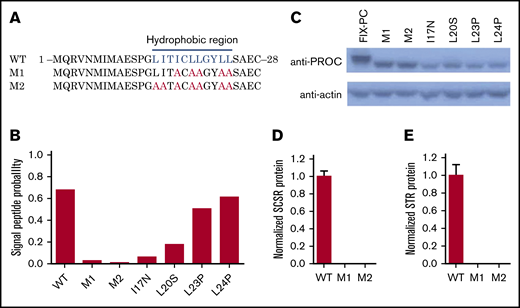
Mutations in the hydrophobic region of the signal peptide block cotranslational translocation of the reporter. (A) The mutated amino acid sequence of FIX signal peptide, as indicated in hydrophobic region. The blue letters indicate the hydrophobic residues in the region, and the red letters indicate the corresponding residues mutated to alanines. (B) SignalP prediction for signal peptide probability of different mutants. (C) Western blot shows that mutations destroying function in the hydrophobic region lead to lower MW. D and E. SCSR (D) and STR (E) proteins are undetectable in M1 and M2. WT, wild-type.
Mutations in the hydrophobic region of the signal peptide block cotranslational translocation of the reporter. (A) The mutated amino acid sequence of FIX signal peptide, as indicated in hydrophobic region. The blue letters indicate the hydrophobic residues in the region, and the red letters indicate the corresponding residues mutated to alanines. (B) SignalP prediction for signal peptide probability of different mutants. (C) Western blot shows that mutations destroying function in the hydrophobic region lead to lower MW. D and E. SCSR (D) and STR (E) proteins are undetectable in M1 and M2. WT, wild-type.
To confirm this hypothesis, the essential hydrophobic residues of the signal peptide were mutated to alanines to ensure the dysfunction of cotranslational translocation in this region (M1 and M2; Figure 3A). The signal peptide prediction also assumes that the mutations eliminate the function of the signal peptide (Figure 3B). As expected, western blot results show a low MW band in M1 and M2 as the 4 missense mutations (Figure 3C). Moreover, the SCSR and STR levels of M1 and M2 are undetectable (Figure 3D-E). Collectively, these results suggest that these mutations lead to FIX deficiency because of the dysfunction of the signal peptide via its interference with cotranslational translocation of FIX into the ER.
Characterization of the protein-maturation process and γ-carboxylation of the FIX-PC reporter
The maturation of FIX in the ER requires several steps, including signal peptide cleavage, propeptide cleavage, γ-carboxylation, and glycosylation.3,25 To assess whether these mutations affect these processes, we analyzed the electrophoresis mobility and γ-carboxylation level of the intracellular FIX-PC reporter.
Because FIX is a vitamin K–dependent protein, the effect of vitamin K on protein maturation and γ-carboxylation was determined first. At least 4 bands were observed in the process of protein maturation using anti–protein C antibody (supplemental Figure 4A; long exposure). Interestingly, the band with the highest MW (band 1), depending on vitamin K, should be one of the states of the γ-carboxylated reporter. By using an anti-Gla antibody, vitamin K is required to detect the γ-carboxylated reporter (supplemental Figure 4A, bottom panel); however, in the absence of vitamin K, we observed nonspecific bands that were at a background (∼20%) level on the immunoblot (supplemental Figure 4B).
Next, we assessed the protein processing and γ-carboxylation of these mutations. By using the anti–protein C antibody, most mutations show a similar band pattern as wild-type FIX-PC, except that A26D and C28R/Y/W in the signal peptide have slower electrophoresis mobility (Figure 4A, upper panel). By using the anti-Gla antibody, the γ-carboxylated reporter is decreased in mutations of A26D, C28W, and A37D (Figure 4A, lower panel). Further, our results show that the relative γ-carboxylation efficiency of most mutations is similar to that of wild-type FIX-PC. However, mutations in A26D, C28W, and A37D show decreased relative γ-carboxylation efficiency; in contrast, mutations in C28Y and V30I show increased relative γ-carboxylation efficiency (Figure 4B). The complexity of protein processing and γ-carboxylation of the mutations suggests that various mechanisms underlie the decrease in the SCSR level. To better understand their potential mechanisms, we address them in detail in the following paragraphs.
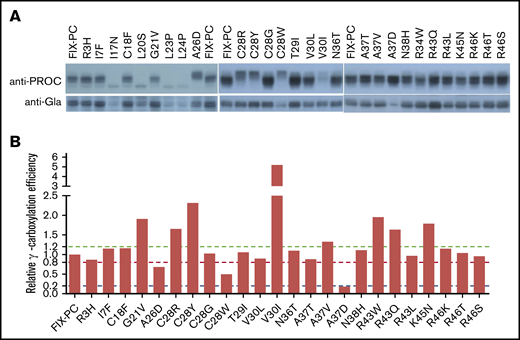
Characterization of intracellular reporter protein maturation and γ-carboxylation of the missense mutations. (A) Electrophoresis mobility assay shows intracellular reporter protein maturation process of the intracellular FIX-PC reporter, as detected by anti–protein C antibody (upper panel). Western blot shows γ-carboxylation of the reporter, as detected by anti-Gla antibody (lower panel). (B) Relative γ-carboxylation efficiency. Western blot bands in (A) were quantified by using ImageJ, and the γ-carboxylated reporter/total intracellular reporter ratio was calculated and normalized to the ratio of wild-type FIX-PC to give the relative γ-carboxylation efficiency. The green, red, and blue dashed lines show 1.2-fold, 0.8-fold, and 0.2-fold relative γ-carboxylation efficiency of wild type, respectively.
Characterization of intracellular reporter protein maturation and γ-carboxylation of the missense mutations. (A) Electrophoresis mobility assay shows intracellular reporter protein maturation process of the intracellular FIX-PC reporter, as detected by anti–protein C antibody (upper panel). Western blot shows γ-carboxylation of the reporter, as detected by anti-Gla antibody (lower panel). (B) Relative γ-carboxylation efficiency. Western blot bands in (A) were quantified by using ImageJ, and the γ-carboxylated reporter/total intracellular reporter ratio was calculated and normalized to the ratio of wild-type FIX-PC to give the relative γ-carboxylation efficiency. The green, red, and blue dashed lines show 1.2-fold, 0.8-fold, and 0.2-fold relative γ-carboxylation efficiency of wild type, respectively.
The residues of C28 and A26 are located in the signal peptidase recognition region, and signal peptide prediction indicates that mutations in both residues decrease the signal peptide probability of FIX (supplemental Figure 5); therefore, the slower electrophoresis mobility indicates that mutations in A26D and C28R/Y/W may affect reporter protein processing by interfering with cleavage of the signal peptide. In addition, our results indicate that these mutations may also affect the γ-carboxylation efficiency of the reporter (Figure 4) by the uncleaved signal peptide, which leads to a longer propeptide to affect the subsequent γ-carboxylation of the Gla domain. In addition, the A26D mutation may affect secretion of the reporter because its intracellular protein expression is not affected (supplemental Figure 3), whereas the SCSR and STR levels are decreased significantly (Figure 2A-B).
Mutation of V30I in the propeptide region decreases protein expression significantly, consistent with the FIX deficiency in patients.29 However, the relative γ-carboxylation efficiency is significantly increased in V30I (Figure 4B), which is consistent with its increase of the normalized RC/T of the secreted reporter (Figure 2C). We conducted a signal peptide prediction for V30I because this residue is close to the signal peptide, but we found that V30I did not affect the signal peptide probability of FIX (supplemental Figure 5). Therefore, an additional study is required to explore how V30I decreases protein expression and increases the relative γ-carboxylation efficiency at the same time.
The 3 types of A37 mutations in the propeptide region have different effects on γ-carboxylation. Our results show that A37D cannot be γ-carboxylated (Figure 4), but it does not affect protein expression (supplemental Figure 3). The SCSR level of A37D was consistently undetectable (Figure 2A). This mutation causes FIX deficiency, probably because it generates a dysfunctional propeptide that leads to an uncarboxylated Gla domain. The other mutations, A37T and A37V, were reported to cause bleeding symptoms during the course of treatment with vitamin K antagonists.30 Our data show that both mutations decrease the SCSR level (Figure 2A), consistent with previous results from Hao et al25 and Pezeshkpoor et al.30 Moreover, our analysis of the normalized RC/T (Figure 2C), the relative γ-carboxylation efficiency (Figure 4B), and the intracellular protein expression level shows that A37T primarily affects γ-carboxylation efficiency, whereas A37V largely affects protein secretion.
The propeptide is cleaved off after γ-carboxylation of the Gla domain. Unremoved propeptide leads to FIX with an extended N terminus that abolishes proper folding of the Gla domain,8,9,20 resulting in FIX dysfunction.7,18 Residues 43 through 46 are essential for propeptide cleavage. Naturally occurring mutations on residues R43 and R46 interfere with propeptide cleavage and lead to FIX with an extended N terminus.6,31 However, it remains controversial whether these mutations affect γ-carboxylation of the Gla domain.6,7,32,33 To resolve this issue, we analyzed all missense mutations on residues 43 through 46. Our results show that these mutations do not affect intracellular expression or γ-carboxylation of the reporter (Figure 4), but they do decrease the SCSR level (Figure 2A). Thus, these mutations only interfere with propeptide cleavage.
Different bleeding tendencies in patients with the same mutation may be caused by different levels of vitamin K in the body
Given that the SCSR level could represent bleeding tendencies, we investigated the correlation between them. Most mutations show a mild or normal decrease in their SCSR levels; only a subset of mutations exhibit moderately (L24P, R43W/Q/L, K45N, R46K/T) or severely (L23P, A37D, and R46S) decreased levels (Figure 2A). As a comparison, most patients reported in the database have moderate or severe bleeding tendencies; very few are mild (supplemental File 1).3 In addition, patients carrying the same mutation in the database have different bleeding tendencies (Figure 5A), an observation that was also implied in recent studies.5,10 To resolve these inconsistencies, we investigated the effect of vitamin K level on SCSR level, because the vitamin K level may vary widely in patients and cause the phenotypic heterogeneity.5 Indeed, our results showed that, with the exception of L23P, L24P, and A37D mutations, the SCSR level depends on the vitamin K concentration (Figure 5B; supplemental Figure 6). Thus, the discrepancy between the SCSR levels in our experiments and the bleeding tendencies in patients is probably due to different levels of vitamin K in the 2 settings. Furthermore, variations in the vitamin K level in patients may explain why the same mutation has different bleeding tendencies in different patients.
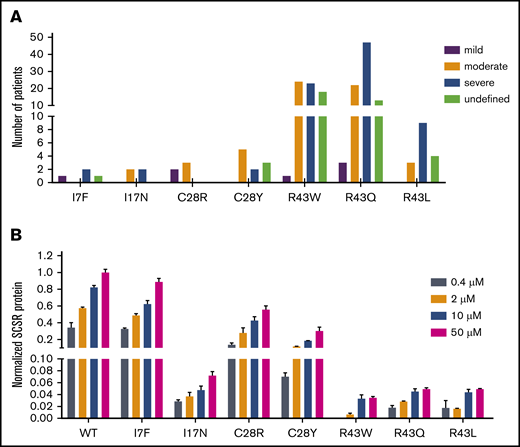
Vitamin K level explains the discrepancy in bleeding tendencies in patients with the same mutation. (A) Different bleeding tendencies were observed in hemophilia B patients with the same mutation in the database. (B) The SCSR levels of the mutations are dependent on the vitamin K concentration. SCSR levels of the mutants were corrected by Metridia luciferase and normalized to the SCSR level of wild-type (WT) FIX-PC at 50 μM vitamin K. The error bars indicate the standard deviation from 3 biological replicates.
Vitamin K level explains the discrepancy in bleeding tendencies in patients with the same mutation. (A) Different bleeding tendencies were observed in hemophilia B patients with the same mutation in the database. (B) The SCSR levels of the mutations are dependent on the vitamin K concentration. SCSR levels of the mutants were corrected by Metridia luciferase and normalized to the SCSR level of wild-type (WT) FIX-PC at 50 μM vitamin K. The error bars indicate the standard deviation from 3 biological replicates.
Discussion
The signal peptide and propeptide of FIX regulate protein secretion and γ-carboxylation, respectively. However, FIX deficiency has not been well understood in hemophilia B patients with mutations in these regions. In this study, we modified a cell-based reporter assay15,25,34,35 and used the FIX-PC reporter to characterize the missense mutations in the signal peptide and propeptide regions of FIX. Our results demonstrate that the SCSR level of most mutations is decreased significantly, which is consistent with FIX deficiency in hemophilia B patients; however, different mechanisms of FIX deficiency are involved in these mutations (Figure 6).

The effect of mutations on functional subdomains of FIX signal peptide and propeptide. The signal peptide consists of 2 subdomains: the hydrophobic region and the signal peptidase recognition site. The hydrophobic region is recognized by signal recognition particles that assist in cotranslational translocation of FIX into the ER; mutations in the region lead to a lower MW. Mutations in the signal peptidase recognition site interfere with protein secretion, expression, and γ-carboxylation efficiency by preventing signal peptide cleavage. The propeptide region includes 2 subdomains: the GGCX recognition site and the propeptidase recognition site. Mutations in the GGCX recognition site affect protein secretion, protein expression, or γ-carboxylation efficiency. Mutations in the propeptidase recognition site lead to unremoved propeptide and destroy the specific conformation of the Gla domain.
The effect of mutations on functional subdomains of FIX signal peptide and propeptide. The signal peptide consists of 2 subdomains: the hydrophobic region and the signal peptidase recognition site. The hydrophobic region is recognized by signal recognition particles that assist in cotranslational translocation of FIX into the ER; mutations in the region lead to a lower MW. Mutations in the signal peptidase recognition site interfere with protein secretion, expression, and γ-carboxylation efficiency by preventing signal peptide cleavage. The propeptide region includes 2 subdomains: the GGCX recognition site and the propeptidase recognition site. Mutations in the GGCX recognition site affect protein secretion, protein expression, or γ-carboxylation efficiency. Mutations in the propeptidase recognition site lead to unremoved propeptide and destroy the specific conformation of the Gla domain.
The signal peptide contains a hydrophobic region and a signal peptidase recognition site (Figure 6). The hydrophobic region helps cotranslational translocation of FIX into the ER, and the signal peptidase recognition site is for signal peptide cleavage. Thus, missense mutations (I17N, L20S, L23P, and L24P) in the hydrophobic region interfere with cotranslational translocation of FIX into the ER, and mutations (A26D and C28R/Y/W) in the signal peptidase recognition site interrupt signal peptide cleavage. Interference with signal peptide cleavage will lead to a longer propeptide, which, in turn, affects protein expression (C28R/Y/W), protein secretion (A26D), and γ-carboxylation of the Gla domain (A26D and C28R/Y/W). It is intriguing that the longer propeptide can decrease or increase γ-carboxylation efficiency, requiring further study to elucidate the underlying mechanism.
The propeptide contains 2 elements: the GGCX recognition site and the propeptidase recognition site25 (Figure 6). This study indicates that various mechanisms are involved in the FIX deficiency in missense mutations of both recognition elements. For mutations in the GGCX recognition site, V30I decreases FIX expression, A37D leads to uncarboxylated FIX, A37T primarily decreases γ-carboxylation efficiency, and A37V primarily affects FIX secretion. These distinct mechanisms indicate that the GGCX recognition site is involved in γ-carboxylation of the Gla domain, as well as in protein expression and secretion. In addition, our study clarifies that mutations in the propeptidase recognition site interfere with propeptide cleavage and prevent proper folding of the Gla domain; however, they do not affect γ-carboxylation of the Gla domain.25
Importantly, our data show that the SCSR level depends on vitamin K concentration in most mutations, suggesting that oral administration of vitamin K may alleviate the severity of bleeding tendencies for patients carrying certain missense mutations in the signal peptide and propeptide. For instance, R43 is a mutation hotspot that accounts for 73.5% of hemophilia B cases; in this study, we find that vitamin K supplementation can improve the SCSR level of R43 mutants from severe (<1%) to moderate or mild (∼5%). Therefore, we assumed that R43 mutations significantly decrease cleavage efficiency between R46 and Y47 by propeptidase, but there is still a little functional FIX, which can well explain why patients carrying R43 mutations have different bleeding tendencies. Although our results suggest that oral administration of vitamin K may be a strategy to alleviate the severity of bleeding tendencies in patients who have missense mutations in the FIX signal peptide and propeptide regions, it remains to be demonstrated in clinical practice.
In this study, our modified cell-based reporter assay provided a reasonable explanation for most hemophilia B patients with missense mutations in the signal peptide and propeptide; however, this method may have some limitations (eg, it may not be applicable to mutations altering DNA transcription or messenger RNA splicing). Signal peptide and propeptide are encoded by exon 1 (1-29) and exon 2 (30-77) of the FIX gene. It had been reported that mutations in exons can change the gene’s splicing site,34,36-38 and those in exon 1 may affect the binding of transcription factors.39 Thus, further study is required to understand how several missense mutations, such as R3H, I7F, C18F, G21V, and T29I, lead to hemophilia B.
Data sharing requests should be sent to Guomin Shen (shenba433@163.com or shenguomin@haust.edu.cn).
Acknowledgments
This work was supported by grants 81770140 (G.S.), 31900412 (M.G.), and 31301135 (H. Yu) from the National Natural Science Foundation of China; grant 182102410079 (G.S.) from the Henan Department of Science and Technology; grant 2017GGJS069 (G.S.) from the Henan Department of Education; and National Institutes of Health grants HL121718 (National Heart Lung, and Blood Institute) and EY028705 (National Eye Institute) (both W.L.).
Authorship
Contribution: G.S. and W.L. designed the study, analyzed the data, and wrote the manuscript with help from Y.C., C.L., and H. Yu; W.G. created all of the constructs with help from Y.W., L.C., R.H., and Y.S.; W.G. performed the enzyme-linked immunosorbent assay assay, with help from S.L.; Y.X. performed and analyzed the western blots with help from M.G., Q.C., and Y.C.; and H.L. collected and analyzed data from the FIX variants database, with help from H. Yang.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Guomin Shen, Institute of Hemostasis and Thrombosis, College of Medicine, Henan University of Science and Technology, No 263 Kaiyuan Ave, Luoyang, Henan 471003, People’s Republic of China; e-mail: shenba433@163.com or shenguomin@haust.edu.cn; and Weikai Li, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, 660 S Euclid Ave, St Louis, MO 63110; e-mail: weikai@wustl.edu.

