

Key Points
We explore the important role of the clinical value of MRD use in the real-world practice in MM.
We address new topics in the field of MRDs, including the importance of MRD dynamics and prediction of relapse.

Abstract
Few clinical studies have reported results of measurable residual disease (MRD) assessments performed as part of routine practice. Herein we present our single-institution experience assessing MRD in 234 multiple myeloma (MM) patients (newly diagnosed [NDMM = 159] and relapsed [RRMM = 75]). We describe the impact of depth, duration, and direction of response on prognosis. MRD assessments were performed by next-generation sequencing of immunoglobulin genes with a sensitivity of 10−6. Those achieving MRD negativity at 10−6, as well as 10−5, had superior median progression-free survival (PFS). In the NDMM cohort, 40% of the patients achieved MRD negativity at 10−6 and 59% at 10−5. Median PFS in the NDMM cohort was superior in those achieving MRD at 10−5 vs <10−5 (PFS: 87 months vs 32 months; P < .001). In the RRMM cohort, 36% achieved MRD negativity at 10−6 and 47% at 10−5. Median PFS was superior for the RRMM achieving MRD at 10−5 vs <10−5 (PFS: 42 months vs 17 months; P < .01). Serial MRD monitoring identified 3 categories of NDMM patients: (A) patients with ≥3 MRD 10−6 negative samples, (B) patients with detectable but continuously declining clonal numbers, and (C) patients with stable or increasing clonal number (≥1 log). PFS was superior in groups A and B vs C (median PFS not reached [NR], NR, 55 respectively; P < .001). This retrospective evaluation of MRD used as part of clinical care validates MRD as an important prognostic marker in NDMM and RRMM and supports its use as an endpoint in future clinical trials as well as for clinical decision making.
Introduction
Measurable (sometimes called minimal) residual disease (MRD) testing in multiple myeloma (MM) is increasingly being used in clinical trials for the assessment of disease response and as a prognostic tool for predicting response duration. Early data have shown that MRD-negative responses result in improved progression-free survival (PFS) and that MRD positivity after treatment is associated with a higher risk of relapse.1,2 Thus, MRD is becoming a relevant surrogate marker for PFS and possibly overall survival (OS) in MM.1,2
As therapy for myeloma improves and more patients achieve a stringent complete response (sCR), it is imperative that we begin to incorporate techniques to better measure depth of response. Two techniques have demonstrated the ability to accurately measure residual clonal plasma cells in the marrow, including next-generation flow cytometry (EUROFLOW) and next-generation sequencing of immunoglobulin genes (NGS; Clonoseq: Adaptive Technologies).3,4 To date, the majority of MRD data have used next-generation flow cytometry or NGS and come from retrospective or subset analyses of patients enrolled in clinical trials. Few studies have analyzed the prognostic impact of MRD in patients treated in a general practice setting.4-8 This report describes the outcomes of newly diagnosed multiple myeloma (NDMM) and refractory relapsed multiple myeloma (RRMM) patients treated in a general practice setting where NGS was used to analyze depth of response. With new the treatment in relapse setting as daratumumab, we could achieve deep response, achieving >20% of MRD-negative cases, and exploration of this in the real-world setting could be of interest.
In addition, there are few studies analyzing the relevance of monitoring MRD sequentially in patients under treatment, all limited by low numbers of patients and small numbers of time points. A small study of 39 patients reported by the Italian MM group was among the first to establish that MRD dynamics could be another relevant prognostic factor.9 Other clinical studies suggested that MRD kinetics are more informative than single time point assessments and may be useful in addressing specific clinical questions.10
A very relevant, but relatively unexplored area of investigation is the understanding of how monitoring the depth of response in individual patients might inform prognosis and potentially be used to guide therapy. A recent study showed that patients achieving a depth of response at a level of 10−3 had a projected PFS of 35 to 45 months, whereas patients with a depth of response to the level of 10−5 had a projected PFS of >80 months.11 This brings into question whether patients who achieve a lesser response (ie, at a level of 10−3) might benefit from a change in therapy and being treated more aggressively in an attempt to reach a target of 10−5, thereby realizing the benefit from the deeper response.
We present here a single institution’s experience assessing MRD in MM patients receiving both frontline and salvage therapy. We describe the outcomes of NDMM and RRMM patients treated in a general practice setting where NGS is used to analyze depth of response. We report the impact of MRD on depth of response, duration of response, and kinetics of response and how it relates to prognosis. Our findings suggest that MRD dynamics may play an important role in future therapeutic decision making.
Patients and methods
Patients
Two hundred thirty-four MM patients (159 NDMM and 75 RRMM) treated at the University of California San Francisco from 2005 to 2018 were included. Major characteristics of the patients are summarized in Tables 1 and 2. Median follow-up from the first MRD assessment was 29 months (range 2 to 73 months). This retrospective study was approved by the University of California San Francisco Institutional Review Board.
Main patient characteristics at diagnosis
| Overall (N = 234) | Newly diagnosed (n = 159) | Second line and subsequent (n = 75) | |
|---|---|---|---|
| Male, n (%) | 142 (61) | 97 (61) | 45 (60) |
| Mean age (standard deviation), y | 58.1 (9.7) | 58.8 (9.8) | 56.4 (9.1) |
| Myeloma type, n (%) | |||
| IgG | 128 (61) | 88 (62) | 40 (58) |
| IgA | 39 (18) | 21 (15) | 18 (26) |
| Light chains | 44 (21) | 33 (23) | 11 (16) |
| High-risk cytogenetics,* n (%) | 40 (21) | 30 (23) | 10 (17) |
| ISS, n (%) | |||
| I | 70 (36) | 52 (39) | 18 (30) |
| II | 53 (27) | 33 (25) | 20 (33) |
| III | 71 (37) | 48 (36) | 23 (38) |
| Overall (N = 234) | Newly diagnosed (n = 159) | Second line and subsequent (n = 75) | |
|---|---|---|---|
| Male, n (%) | 142 (61) | 97 (61) | 45 (60) |
| Mean age (standard deviation), y | 58.1 (9.7) | 58.8 (9.8) | 56.4 (9.1) |
| Myeloma type, n (%) | |||
| IgG | 128 (61) | 88 (62) | 40 (58) |
| IgA | 39 (18) | 21 (15) | 18 (26) |
| Light chains | 44 (21) | 33 (23) | 11 (16) |
| High-risk cytogenetics,* n (%) | 40 (21) | 30 (23) | 10 (17) |
| ISS, n (%) | |||
| I | 70 (36) | 52 (39) | 18 (30) |
| II | 53 (27) | 33 (25) | 20 (33) |
| III | 71 (37) | 48 (36) | 23 (38) |
ISS, International Scoring System.
High-risk cytogenetics was defined as del 17p; t (4;14), t(14;16), or t(14;20).
Summary of the treatment of newly diagnosed patients
| n (%) | |
|---|---|
| Induction | |
| CyBorD | 42 (26) |
| RVD | 80 (50) |
| Others | 37 (23) |
| ASCT, yes | 136 (86) |
| Double ASCT, yes | 5 (4) |
| Consolidation post ASCT, yes | 17 (11) |
| Maintenance, yes | 135 (85) |
| Type of maintenance | |
| Lenalidomide | 69 (51) |
| Proteasome inhibitors | 20 (15) |
| IMiD+ proteasome inhibitors | 29 (22) |
| Others | 16 (12) |
| n (%) | |
|---|---|
| Induction | |
| CyBorD | 42 (26) |
| RVD | 80 (50) |
| Others | 37 (23) |
| ASCT, yes | 136 (86) |
| Double ASCT, yes | 5 (4) |
| Consolidation post ASCT, yes | 17 (11) |
| Maintenance, yes | 135 (85) |
| Type of maintenance | |
| Lenalidomide | 69 (51) |
| Proteasome inhibitors | 20 (15) |
| IMiD+ proteasome inhibitors | 29 (22) |
| Others | 16 (12) |
ASCT, autologous stem cell transplantation; CyBorD, bortezomib, cyclophosphamide, dexamethasone; IMiD, immunomodulatory drugs; RVD, bortezomib, revlimid, dexamethasone.
Treatment
Patients received anti-MM therapy per provider preference with the aim of obtaining maximal response by International Myeloma Working Group (IMWG) criteria.12 Newly diagnosed patients generally received induction with triplet combinations, which consisted of proteasome inhibitors, immunomodulators (or alkylators), and corticosteroids and then proceeded to ASCT followed by maintenance therapy. In summary, 86% of patients received ASCT, 11% of patients received consolidation, and 84% of patients received maintenance therapy until relapse or unacceptable toxicity. MRD was assessed, in general, when patients achieved complete response (CR; 98%), although a few of them were considered very good partial response (2%). There was no uniform timing of MRD assessments with the majority of patients being tested within the first year after ASCT.
Among the RRMM patients who had MRD testing, 54% were in the second line, 29% were in the third line, and 17% were in the fourth line or later. These patients received a variety of regimens according to physician preference and continued therapy until disease progression or unacceptable toxicity. MRD assessments were performed in select patients who achieved CR with no uniform timing of MRD assessment.
MRD assessment
Fresh or stored bone marrow samples were sent to Adaptive Biotechnologies (Clonoseq; Seattle, WA), and patient-specific clonal rearrangements were identified via NGS of immunoglobulin genes (IGH-VDJH and IGK or IGH-VDJH, IGH-DJH, IGK, and IGL). Patients for which unique sequences were not identified were excluded from MRD analysis. For MRD quantification, fresh bone marrow samples were obtained, and DNA was isolated, amplified by polymerase chain reactions using immunoglobulin gene-specific primers, and sequenced. Once the absolute amount of total cancer-derived molecules present in a sample was determined, a final MRD measurement was calculated, and the number of cancer-derived molecules per 1 million cell equivalents was provided. In cases where 2 or more tumor clones existed, the clone with the highest MRD value was reported.
Although the sensitivity of this technique ranged from 10−4 to 10−6, 96% of the samples reported data at the level of 10−6. These levels of sensitivity were based on the DNA availability of each sample. MRD levels were reported as follows: <10−6; 10−6 to <10−5; 10−5 to <10−4; and ≥10−4.
As noted above, MRD samples were obtained from patients achieving complete remission following antimyeloma therapy. In the NDMM patients, 1 MRD assessment was obtained in 68 patients, 2 to 3 samples were obtained in 30 patients, and >3 samples were obtained in 61 patients. For RRMM, serial samples were obtained based on provider preference, and no specific protocol was followed.
Statistical analysis
All data were entered into a Redcap database (Vanderbilt University, Nashville, TN). The time of origin for PFS was defined as the date of transplant for newly diagnosed transplanted patients and the time of the first MRD measurement for all other patients. PFS was calculated from the date of transplant for those received transplant and was set to be the date of first MRD test for others to disease progression or death, whichever occurred first. For patients who were alive and free of progression at the end of follow-up, PFS was censored at the date of last disease evaluation. OS was calculated from the time of origin to death. OS for patients without a recorded death date was censored at their last contact date.
PFS and OS were summarized using the Kaplan-Meier method and compared between groups using the log-rank test. Cox regressions were used to evaluate the covariate effects on PFS. Multivariate analysis was performed using an adjusted stepwise Cox proportional regression hazard model. Two-sided P < .05 was considered statistically significant. The statistical analysis was performed with R language (version 3.6.1, R Foundation for Statistical Computing, Vienna, Austria).
Results
Sequence identification
Successful identification of >1 trackable sequence in the pretreatment sample occurred in 234 out of 251 (93%) patients. The immunoglobulin genes used for MRD assessment were as follows: IGH DJ (73 cases; 31%), IGH VDJ (74 cases; 32%), IGK VJ (84 cases; 36%), and IGL VJ (3 cases; 1%). There was no difference in survival when comparing the genes studied for MRD assessment.
Proportion of patients achieving MRD negativity on at least 1 occasion (Table 3).
Overall population
A total of 525 MRD samples from 234 patients were analyzed at various time points during their disease course. MRD data were available at ≥3 time points for 73 patients, 2 time points for 49 patients, and 1 time point for 112 patients. Overall, 91 of 234 patients (39%) achieved MRD negativity at 10−6, and 129 (55%) achieved MRD at ≤10−5 on 1 or multiple assessments.
Newly diagnosed population
In the NDMM group (n = 159), the median PFS from diagnosis was 86.4 months. Event-free probability at 5 years was 59.5% for PFS and 93% for OS. In this NDMM group, 64 of 159 (40%) patients achieved undetectable MRD at 10−6 on at least 1 occasion. These patients had a prolonged PFS in comparison with patients who were persistently MRD positive (87 months vs 71 months; P < .001; hazard ratio [HR] 2.49; 95% confidence interval [CI], 1.34 to 4.62; Figure 1A). The 3-year survival probability for patients with MRD negative was 90% (95% CI, 81% to 98.78%) and 56.6% (95% CI, 45.7% to 70.1%) for patients with MRD positive (P < .001). When using the IMWG definition of MRD negativity (<10−5), 94 out of 159 (59%) patients achieved MRD negativity, and these patients had a prolonged PFS in comparison with those who were persistently MRD positive (87 months vs 32 months; P < .001; HR 3.54; 95% CI, 1.94 to 6.45; Figure 1B). The 3-year survival probability for patients with MRD <10−5 was 85.9% (95% CI, 78.2% to 94.5%) and 46.8% (95% CI, 33.9% to 64.7%) for patients with MRD >10−5 (P < .001). The median follow-up for death was 39.4 months; only 2 out of 64 patients achieving MRD negativity had died.
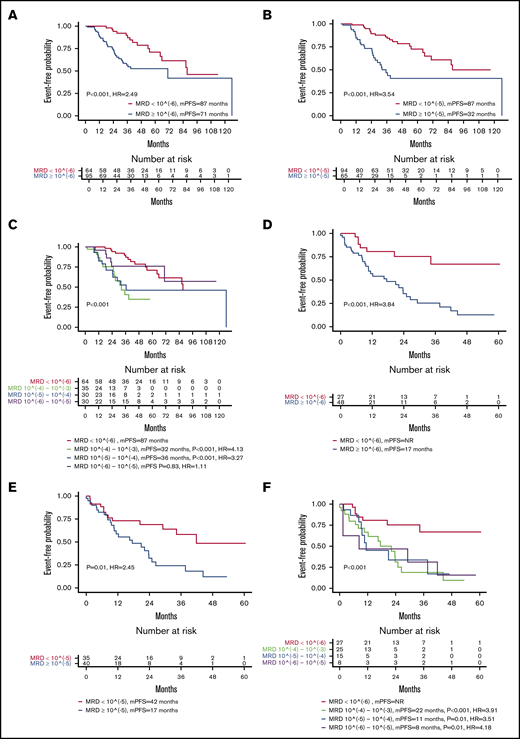
Impact on PFS of MRD levels. Kaplan-Meier curves for PFS in newly diagnosed patients achieving MRD <10−6 vs ≥10−6 (A); newly diagnosed patients achieving MRD <10−5 vs ≥10−5 (B); newly diagnosed patients classified according to depth of response in 4 MRD categories (<10−6, 10−6 to 10−5, 10−5 to 10−4, 10−4 to 10−3) (C); patients on second-line or later therapy achieving MRD <10−6 vs ≥10−6 (D); patients on second-line or later therapy achieving MRD <10−5 vs ≥10−5 (E); and patients on second-line or later therapy achieving MRD <10−6 vs ≥10−6 (F). mPFS, median progression-free survival.
Impact on PFS of MRD levels. Kaplan-Meier curves for PFS in newly diagnosed patients achieving MRD <10−6 vs ≥10−6 (A); newly diagnosed patients achieving MRD <10−5 vs ≥10−5 (B); newly diagnosed patients classified according to depth of response in 4 MRD categories (<10−6, 10−6 to 10−5, 10−5 to 10−4, 10−4 to 10−3) (C); patients on second-line or later therapy achieving MRD <10−6 vs ≥10−6 (D); patients on second-line or later therapy achieving MRD <10−5 vs ≥10−5 (E); and patients on second-line or later therapy achieving MRD <10−6 vs ≥10−6 (F). mPFS, median progression-free survival.
Multivariable analysis using the stepwise variable selection procedure selected 4 predictors: MRD (>10−6 vs <10−6), age at diagnosis (decade), myeloma type, and ISS. Only MRD > 10−6 vs MRD < 10−6. HR 3.11 (1.41, 6.87) (P < .001); age at diagnosis (decade) HR 1.63 (1.05, 2.52) (P < .03); and MM type (IgG vs IgA) HR 0.3 (0.12, 0.78) (P < .01).
Interestingly, in 30 NDMM patients with high-risk cytogenetic features, MRD negativity at 10−6 (n = 10, 33%) was able to identify patients with longer PFS (median PFS not reached [NR] vs NR, P < .04). The 3-year survival probability for patients with MRD < 10−6 was 100% and 64.1% (95% CI, 44.1% to 93.1%) for patients with MRD ≥ 106 (P < .001).
Furthermore, patients who were MRD negative at 10−6 or who were MRD positive at a very low level (between 10−5 and 10−6) had a better PFS than those with higher disease burdens (>10−5) (P < .001; Figure 1C).
Relapsed population
Overall, 27 of 75 (36%) patients achieved undetectable MRD at 10−6 on at least 1 occasion. These patients had a prolonged PFS in comparison with patients who were persistently MRD positive at different levels (NR vs 17 months; P < .001; HR, 3.84; 95% CI, 1.69 to 8.73; Figure 1D,F). Moreover, 35 of the 75 achieved MRD at <10−5 (47%), and PFS was prolonged in these patients vs patients who did not achieve negative status at a level <10−5 status (42 months vs 17 months; P = .01; HR, 2.45; 95% CI, 1.25 to 4.82; Figure 1E). The median follow-up time for death is 26.3 months (range 2.5 to 69.8); a total of 9 deaths were observed, and no differences were observed between MRD groups.
In the multivariable analysis using the stepwise variable selection procedure, the only predictor selected was MRD (>10−6 vs <10−6), HR 3.84 (1.69 to 8.73) (P < .001).
MRD monitoring and dynamics
We then analyzed the effect of repeated MRD monitoring on PFS in 61 newly diagnosed patients who had ≥3 MRD assessments. Three categories were identified in newly diagnosed patients: (A) patients with ≥3 MRD-negative measurements at 10−6 (n = 21), (B) patients with continuously declining detectable clones (≥1 log: n = 19), and (C) patients with a stable or growing number of clones (≥1 log: n = 21). Groups A and B had a more prolonged PFS than group C (NR vs NR vs 55 months; P < .001; Figure 2A).
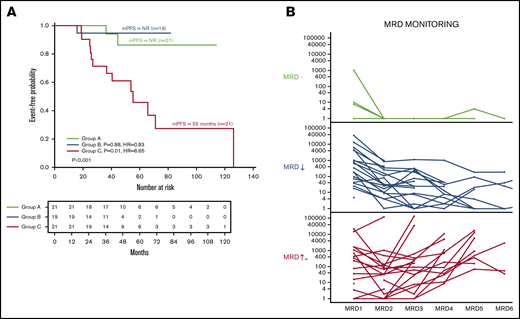
MRD dynamics and its impact on PFS. (A) Kaplan-Meier curves for PFS in patients classified according to dynamics of response: group A, patients with 3 or greater MRD-negative assessments (n = 21); group B, patients with continuously declining detectable clones in success time points (n = 19); and group C, patients with stable number of clones (n = 21). (B) MRD evolution for each individual patient by MRD dynamics.
MRD dynamics and its impact on PFS. (A) Kaplan-Meier curves for PFS in patients classified according to dynamics of response: group A, patients with 3 or greater MRD-negative assessments (n = 21); group B, patients with continuously declining detectable clones in success time points (n = 19); and group C, patients with stable number of clones (n = 21). (B) MRD evolution for each individual patient by MRD dynamics.
Interestingly, when we defined a molecular relapse by a rise of >1 logarithm, serial MRD testing was able to predict clinical relapse in 9 out of 10 cases, 4 of which had a confirmatory MRD sample. Molecular relapse preceded a relapse as defined by conventional IMWG standards at a median of 13 months (range 1 to 28 months). Of note, with this technology, we were not able to detect any newly emerging clones in patients with serial MRD assessments during the time of follow-up.
Discussion
Over the last 2 decades, there has been enormous progress in the treatment of MM. With the introduction of novel agents, such as immunomodulatory drugs, proteasome inhibitors, monoclonal antibodies, and chimeric antigen receptor T cells, an sCR can now be achieved in a significant proportion of NDMM and RRMM patients.6,13-15 Consequently, more sensitive assays for detecting and monitoring clinically meaningful residual disease have become relevant tools for assessing response and estimating prognosis. MRD measurements have the ability to accurately predict PFS and OS in large populations, and to evaluate the comparative efficacy of novel therapies. However, the application of MRD testing for prognosis or on making clinical decisions in individual patients treated in routine practice has not been reported.
The data presented in this paper support the role of MRD assessment in the prediction of survival, corroborating the findings of both prospective trials and retrospective analysis performed by other groups. These data add to the growing field of MRD outcomes research and suggest that MRD assessment might be used for real-time clinical evaluation and may be ready for integration into clinical decision making.
MRD detection by NGS of immunoglobulin genes is feasible, and our single-center data have similar applicability (93%) in clinical practice to previous reports from clinical trials.4,5,8 A possible therapeutic strategy therefore in newly diagnosed MM patients could be to obtain a higher proportion of deep molecular responses using MRD as a guide (in our NDMM series, this resulted in 59% of our patients achieving MRD negativity at 10−5). This translated into an estimated PFS in newly diagnosed patients of 86.4 months. This PFS is considerably longer than data reported previously for newly diagnosed patients (50 months for patients achieving very good partial response/CR).1,16,17 This suggests that an MRD-driven decision-making strategy may be successful in improving outcomes in newly diagnosed patients. This, however, must be confirmed in future prospective studies.
Furthermore, this intensifies the debate on the true value of CR and sCR without MRD data in MM. Based on our results, as well as others, we propose that the therapeutic objective in NDMM should be to obtain a sustained deep response as measured by MRD, regardless of therapy and IMWG.
The threshold to determine MRD negativity has been established by IMWG criteria as 10−5,12 although some authors have argued for 10−6.4 In our series, patients who obtained a molecular response at 10−5 had similar outcomes than those achieving MRD negativity at 10−6. This could be explained by the dynamics of MRD and the possibility that the patients did achieve MRD negativity at 10−6 at another time point. We have found several patients receiving maintenance treatment with 10−5 on several MRD assessments who had a similar prognosis to those patients who achieved 10−6 on just 1 MRD measurement. This supports the relevance of sustained MRD negativity at a level of 10−5.
The current IMWG definitions of response ignore depths of response between sCR and MRD negative at 10−5. Instead, we propose the adaptation of a model already in use for other hematologic diseases (such as chronic myeloid leukemia) in which MRD responses are stratified according to the level of tumor depletion, without considering the technique employed. Table 4 presents a proposal in line with this concept, which will have to be further studied to determine its applicability. Our proposed new definition of MRD response includes different MRD levels and will be more useful for comparing the efficacy of different treatment strategies, and for implementing individualized therapy-monitoring strategies. This definition will be applicable in all clinical settings and interchangeable between different centers.
Classification of the response based on depth of response
| MRD response criteria | Abbreviation | Level of reduction |
|---|---|---|
| MRD grade 3 | MRD3 | Nonclonal plasma cells or clonotypes between 10−3 and 10−4 |
| MRD grade 4 | MRD4 | Nonclonal plasma cells or clonotypes between 10−4 and 10−5 |
| MRD grade 5 | MRD5 | Nonclonal plasma cells or clonotypes between 10−5 and 10−6 |
| MRD grade 6 | MRD6 | Nonclonal plasma cells or clonotypes <10−6 |
| MRD response criteria | Abbreviation | Level of reduction |
|---|---|---|
| MRD grade 3 | MRD3 | Nonclonal plasma cells or clonotypes between 10−3 and 10−4 |
| MRD grade 4 | MRD4 | Nonclonal plasma cells or clonotypes between 10−4 and 10−5 |
| MRD grade 5 | MRD5 | Nonclonal plasma cells or clonotypes between 10−5 and 10−6 |
| MRD grade 6 | MRD6 | Nonclonal plasma cells or clonotypes <10−6 |
There are very few studies addressing the serial assessment of MRD in MM, although this strategy has been successfully used in other diseases, such as lymphoma or leukemia,18-20 even knowing the stability of VDJ along the natural history of MM.21 Here, we have demonstrated that MRD monitoring by NGS is feasible and provides reproducible data, as shown in Figure 2B.
Interestingly, we also found a select group of patients in which MRD assessment could predict clinical relapse, anticipating it almost a year earlier. Although the number of patients is small, this adds to the growing call for assessing molecular relapse in MM, and if confirmed with additional studies, may allow us to make earlier clinical decisions. This new concept of molecular relapse should be incorporated in the future in the criteria of relapse of MM. It should be defined as the increment of 1 logarithm of clonotypes respective to the previous MRD assessment.
The main limitations of this paper are its retrospective nature, the heterogeneity of patient treatments, the different time points of the MRD assessments, and the limited number of patients followed for MRD dynamics; however, we believe these limitations are addressed by the nature of this retrospective review and the large number of samples analyzed.
Future directions in this field are peripheral blood molecular tracking, immuno positron emission tomography–computed tomography with new tracers and new high-throughput mass spectrometry techniques for M-component detection. However, as with bone marrow MRD measurements, we will need well-designed studies to get to a point where we can make clinical decisions based on each of these newer methods of MRD detection.
However, questions remain before MRD testing can be routinely adopted into mainstream practice for MM. In particular, further standardization regarding predictive cutoff values and identification of the most informative time points for testing is still necessary.
In conclusion, MRD is an important predictor of PFS in NDMM and RRMM patients. This study shows that MRD assessment in a clinical practice setting likely has the same predictive power as that seen in clinical trials. Depth of response and MRD dynamics may eventually be employed to predict disease evolution and ultimately could drive clinical decision making, potentially improving the outcome of MM patients.
This study supports the relevance of MRD assessment in real-world clinical practice and the importance of MRD monitoring, and most importantly, establishes the importance of the concept of molecular response and molecular relapse. We believe that MRD assessment should be incorporated into clinical practice and look forward to prospective validation of this.
Data from this paper may be acquired by contacting the corresponding author, Joaquin Martinez-Lopez, at jmarti01@med.ucm.es.
Acknowledgments
This work was supported by grants from Sociedad Española de Hematologia y Hemoterapia and the Cancer Research Innovation Spain Foundation (J.M.-L.).
Authorship
Contribution: J.M.-L., J.W., and T.M. assisted with the study design; S.W.W., N.S., N.B., K.Z., T.M., and J.W. were the study investigators; Y.S. and C.-Y.H. performed data analysis; J.M.-L., J.W., S.W.W., N.S., and T.M. interpreted the data; J.W. and J.M.-L. prepared the manuscript; J.M.-L., J.W., N.S., S.W.W., and T.M. reviewed and revised the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: J.M.-L. is a member of the speakers bureau of Adaptive and owns shares in Altum Sequencing. J.W. is a consultant for Adaptive, Celgene, Amgen, and Teneobio. N.S. recieved research funding from Celgene/BMS, Janssen, Bluebird Bio, Sutro Biopharma, and Teneobio; and held advisory roles for GSK, Amgen, Indapta Therapeutics, Sanofi, BMS, CareDx, Kite, and Karyopharm. The remaining authors declare no competing financial interests.
Correspondence: Joaquin Martinez-Lopez, Hospital Universitario 12 de Octubre, Av Córdoba, s/n Hospital 12 de Octubre, CAA 3°D, 28041 Madrid, Spain; e-mail: jmarti01@med.ucm.es.

