

Key Points
Pembrolizumab yielded only 1 transient response in 12 patients with ALL and MRD.
The 1 response occurred after failure of allogeneic transplant and chimeric antigen receptor T cells, suggesting a mechanism of resistance.

Abstract
The presence of measurable residual disease (MRD) in acute lymphoblastic leukemia (ALL) confers a poor prognosis. CD19-targeted immunotherapy is effective against MRD but is logistically challenging, potentially toxic, and not applicable to T-cell ALL. We thus hypothesized that inhibition of PD-1 with pembrolizumab could also be effective for MRD, but without lineage restriction. The primary objective of this phase 2 study was to evaluate the efficacy of pembrolizumab in patients with ALL and MRD. Key eligibility criteria included adults with B- or T-cell ALL and MRD detectable by multiparameter flow cytometry or quantitative polymerase chain reaction from bone marrow aspirate (BMA) despite chemotherapy (plus ABL kinase inhibitor if Philadelphia chromosome positive). Pembrolizumab 200 mg IV was given every 3 weeks. Response was assessed by BMA using methods that previously detected MRD. The primary end point was complete MRD response rate. We stopped enrollment early; only 1 of 12 (8%) experienced a complete MRD response, which lasted 3 weeks. Interestingly, this patient had previously received hematopoietic cell transplantation and CD19-targeted chimeric antigen receptor–modified T-cell therapy and was the only patient to experience an immune-related adverse event from pembrolizumab (grade 3 Stevens-Johnson syndrome). Median overall survival from enrollment was 12.7 months. In summary, pembrolizumab had minimal activity against MRD but was generally well tolerated. These data can be compared with ongoing anti-PD-1 combination studies in ALL, and they further establish the role of trials specifically for patients with MRD. This trial was registered at www.clinicaltrials.gov as #NCT02767934.
Introduction
The presence of measurable residual disease (MRD) (either as persistence during therapy or reappearance afterward) confers a poor prognosis, as it almost inevitably heralds frank hematologic relapse without additional intervention.1,2 Our center’s experience and that of others also demonstrate the increased relapse risk associated with MRD in the context of allogeneic hematopoietic cell transplantation (HCT).3,4 Unfortunately, for those that have MRD, little is known about the optimal management of this high-risk scenario. However, elimination of MRD is critical to achieve long-term disease control.
Patients who have persistent or re-emergent MRD after cytotoxic chemotherapy are unlikely to derive significant benefit from additional chemotherapy. Therefore, immunotherapy is a particularly attractive approach to this problem, particularly since the disease burden is very low. Proof of this principle has been demonstrated with the CD3-CD19 bispecific T-cell engager blinatumomab.5 Comparable results against MRD have been observed with CD19-targeted chimeric antigen receptor (CAR)–modified T cells for B-cell acute lymphoblastic leukemia (ALL).6,7 Unfortunately, both of these strategies are logistically complex, can cause significant toxicity (including serious neurologic side effects and cytokine release syndrome [CRS]), and are expected to only have activity against CD19+ B-cell ALL. Consequently, some patients with MRD may not be able to receive either.
While blinatumomab and CAR-T cells were still being investigated, we hypothesized that immune checkpoint blockade through inhibition of the PD-1/PD-L1 axis could also provide a benefit for this very high-risk clinical scenario. The anti-PD-1 antibodies pembrolizumab and nivolumab have demonstrated efficacy in several relapsed/refractory B and T lymphoid malignancies with relatively low toxicity, as well as a significantly easier mode of administration than blinatumomab or CAR-T cells.8-10 Based on the proven ability of immunotherapy to eliminate MRD in ALL, the belief that such elimination will translate into improved long-term outcomes in ALL, as well as a strong need for new treatments for this challenging disease, we performed a study of single-agent pembrolizumab for the treatment of MRD in adults with ALL. If this agent proved efficacious for MRD, then it could create a novel method of treatment. It could also provide a rationale to test this drug alone or in combinations as consolidation for patients in complete remission or for those with morphologic relapse or refractory ALL.
Methods
Patient eligibility
Patients were eligible to enroll if they were at least 18 years old with a diagnosis of ALL with MRD, defined as <5% blasts in the bone marrow by morphologic assessment and no clinically apparent extramedullary disease but with quantifiably measurable disease assessed by either multiparameter flow cytometry (MFC) or quantitative reverse-transcriptase polymerase chain reaction (PCR). MRD must have been detected under one of the following circumstances: persistence ≥11 weeks after the start of initial therapy (chosen due to the prognostic significance of persistent MRD 3 months into initial therapy), persistence ≥2 weeks after the start of salvage therapy, or reappearance at any time. They must also have previously received, been ineligible for, or declined treatment with blinatumomab. Patients with Philadelphia chromosome–positive (Ph+) disease must have previously received ≥1 ABL kinase inhibitor. Additional inclusion criteria included adequate organ function and an Eastern Cooperative Oncology Group performance status of 0 or 1.
Patients were not eligible to enroll if they had a known diagnosis of primary immunodeficiency or were receiving systemic steroid therapy or any other form of immunosuppressive therapy within 7 days prior to the first dose of pembrolizumab. We initially excluded patients with prior HCT or other forms of cellular immunotherapy, such as CAR-T cells. However, experience with PD-1 inhibitors in these contexts accumulated during the initial conduct of this trial. Thus, we amended the protocol after the first 9 patients enrolled to include these patients under strict criteria: MRD must have been detected at least 21 days from stem cell infusion or at least 28 days from infusion of other cellular immunotherapy; no active graft-versus-host disease, no immunosuppressive therapy within 7 days of the first dose of pembrolizumab, and ≤10 mg of prednisone (or equivalent) daily; and complete resolution of any antecedent manifestations of CRS or neurologic toxicity attributable to cellular immunotherapy. Other exclusion criteria included prior monoclonal antibody therapy within 4 weeks of the first dose of pembrolizumab; prior chemotherapy, targeted small-molecule therapy, or radiation therapy within 2 weeks; insufficient recovery from prior toxicities from prior treatments (ie, grade ≤1 or at baseline); active central nervous system involvement by ALL; infection requiring systemic therapy; autoimmune disease requiring treatment in the past 2 years; known history of noninfectious pneumonitis; or prior therapy with any immune checkpoint inhibitor.
This study was approved by the University of Washington/Fred Hutchinson Cancer Research Center Cancer Consortium Institutional Review Board (protocol #9458), and all patients gave written informed consent. R.D.C. and P.A.S. performed the data analyses, and all authors had access to primary clinical trial data. The study was registered at www.clinicaltrials.gov as #NCT02767934.
Study treatment and assessments
Pembrolizumab was given at a fixed dose of 200 mg IV every 3 weeks (±3 days). Patients were evaluated for toxicity prior to each dose, and adverse events (AEs) were defined and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (version 5.0).11 In addition, for patients previously treated with immune effector cells or HCT, consensus criteria were used to grade events consistent with CRS, immune effector cell-associated neurotoxicity syndrome, and acute GVHD.12,13 Response was determined by bone marrow examination performed after cycles 1, 2, 4, 6, and 8 (ie, 2-4 days prior to the next planned dose of pembrolizumab), with subsequent assessments left to the discretion of the treating physician. At a minimum, bone marrow exams included an evaluation of morphology as well as a repeat analysis of whatever study (or studies) detected MRD at the time of enrollment (eg, if MRD was detected by MFC and PCR at enrollment, follow-up marrow exams included both MFC and PCR); other studies were included as clinically indicated. Patients were eligible to continue in the absence of morphologic relapse (ie, >5% blasts by morphology), development of extramedullary disease, and prohibitive toxicity, but study treatment could be stopped at the discretion of the patient or treating physician for inadequate response (eg, persistent or rising MRD). Morphologic and MRD response were categorized according to standardized criteria.14,15 Patient enrollment occurred between 1/31/2017 and 6/4/2019, and the date of data cutoff for this analysis was 12/17/2019.
As close as feasible to the timing of these marrow examinations at screening, post-cycle 2, post-cycle 4, and end-of-treatment, peripheral blood also was collected for additional correlative studies. Flow cytometry was performed to assess for changes in circulating T-cell subsets and other markers of cellular immunologic response using techniques previously described.16 Further, serum cytokine and inflammatory marker concentrations were measured by the Luminex assay (Luminex Corporation, Austin, TX) according to the manufacturer’s instructions. Selected patients who had previously received CD19 CAR-T cells had testing of peripheral blood for CAR-T-cell persistence via quantitative PCR for the CAR transgene, as described elsewhere.7
Statistical design
The primary objective of this study was to evaluate the efficacy of pembrolizumab in patients with ALL and MRD. Therefore, the primary end point of this phase 2 trial was the rate of complete MRD response, defined as the percentage of evaluable subjects who achieved a complete MRD response per the standardized criteria cited above. Prespecified secondary objectives included description of the toxicity profile of pembrolizumab in this patient population and a preliminary assessment of how MRD response translated into relapse-free and overall survival.
The design of this trial was inspired by the initial study of blinatumomab for chemorefractory MRD, which assumed a historical rate of complete MRD response to standard chemotherapy of 5%.17 Considering the fact that our study of pembrolizumab enrolled patients that previously received or were ineligible for treatment with blinatumomab, and because this trial began before the approval of blinatumomab for treating MRD, we feel it is reasonable to assume a similar historical rate of complete MRD response for subjects in this trial.
The design was based on Simon’s 2-stage minimax design with a historical response rate of 5% and an assumed-true response rate of 20% for patients receiving pembrolizumab on this study. The design also assumed a 10% type-I error rate and a 20% type-II error rate. Under these assumptions and parameters, the first stage enrolled 12 evaluable patients. If 0 responses were seen among these 12, then the study was to be paused for futility to consider alternative strategies to improve efficacy versus consideration of study closure (the probability of 0 responses among 12 under the alternative hypothesis of 20% response is 0.07). On the other hand, if at least 1 response was seen among the first 12 patients, an additional 9 were to be enrolled for a total of 21. If at least 3 responses occurred among the 21 (≥14.3%), this was to be considered sufficient evidence to conclude that the observed response rate is statistically better than the fixed historical rate of 5%. This yielded a type-I error rate of 0.08 and power of 80% along with an expected sample size of 16.1. Additionally, we devised a stopping rule in the event of unacceptably high rate of early morphologic relapse (ie, >50%).
Results
Patient characteristics
We enrolled 12 patients on this study. Characteristics of interest are summarized in Table 1. Ten patients were enrolled with measurable disease by MFC, 2 of whom also had MRD by PCR. Two patients were enrolled with disease only detectable by PCR (ie, no disease detectable by MFC). By MFC, the median level of disease detected at baseline was 0.054% (range, 0% to 1.1%); among the 2 patients with undetectable disease by MFC, BCR-ABL1 PCR was detectable at 0.01% and 0.09%. Median age was 52.5 years (range, 22-75); median number of prior lines of therapy was 1.5 (range, 1-7), with 50% of patients receiving at least 2 lines of prior therapy. Three patients received prior CAR-T cells (7, 28, and 52 months prior to pembrolizumab), and 3 patients underwent a prior HCT (17, 71, and 86 months prior to pembrolizumab, respectively). No patients had received blinatumomab previously. Aside from those with T-cell ALL (n = 2) and CD19− B-cell ALL (n = 1), the remaining patients were offered blinatumomab for treatment of MRD but declined this due to concerns about toxicity, method of administration, or a combination thereof.
Baseline demographics and disease characteristics
| Characteristic | Patients (N = 12) |
|---|---|
| Age at time of consent, y | |
| Median (range) | 52.5 (22-75) |
| >60 | 5 (42) |
| Level of disease at time of consent, median (range), % | |
| MFC | 0.054 (0-1.1) |
| PCR only | 0.05 (0.01-0.09) |
| Method used for MRD detection at enrollment | |
| MFC only | 8 (67) |
| PCR only | 2 (17) |
| MFC and PCR | 2 (17) |
| B lineage | 10 (83) |
| Ph+ | 6 (50) |
| P190 | 4 (67) |
| P210 | 2 (33) |
| Ph− | 6 (50) |
| Normal cytogenetics | 5 (83) |
| Complex cytogenetics | 1 (17) |
| No. of prior therapies | |
| Median (range) | 1.5 (1-7) |
| 1 | 6 (50) |
| ≥2 | 6 (50) |
| No. with prior HCT | 3 (25) |
| No. with prior CAR T cells | 3 (25) |
| Characteristic | Patients (N = 12) |
|---|---|
| Age at time of consent, y | |
| Median (range) | 52.5 (22-75) |
| >60 | 5 (42) |
| Level of disease at time of consent, median (range), % | |
| MFC | 0.054 (0-1.1) |
| PCR only | 0.05 (0.01-0.09) |
| Method used for MRD detection at enrollment | |
| MFC only | 8 (67) |
| PCR only | 2 (17) |
| MFC and PCR | 2 (17) |
| B lineage | 10 (83) |
| Ph+ | 6 (50) |
| P190 | 4 (67) |
| P210 | 2 (33) |
| Ph− | 6 (50) |
| Normal cytogenetics | 5 (83) |
| Complex cytogenetics | 1 (17) |
| No. of prior therapies | |
| Median (range) | 1.5 (1-7) |
| 1 | 6 (50) |
| ≥2 | 6 (50) |
| No. with prior HCT | 3 (25) |
| No. with prior CAR T cells | 3 (25) |
Values are reported as n (%) of patients unless otherwise indicated.
Efficacy and toxicity of pembrolizumab in MRD-positive ALL
Median number of cycles of pembrolizumab received on study was 2 (range, 1-3). One patient (8%; 95% confidence interval, 1% to 35%) had a complete MRD response by both MFC and PCR after 1 cycle; however, this patient had MRD reappearance by both MFC and PCR after a second cycle of pembrolizumab. This patient also experienced grade 3 Stevens-Johnson syndrome (further details below). Notably, their treatment history included haploidentical allogeneic HCT 5 years earlier and CD19-targeted CAR-T cells 2 years before enrollment in this study. Chimerism testing had not been performed since ∼4 weeks after CAR-T cell treatment (ie, ∼2 years prior to enrollment), at which point they had 100% engraftment in both CD3+ and CD33+ subsets from peripheral blood.
The remaining 11 patients had MRD increase (Figure 1). The changes in disease burden over time appear to increase consistently, though the degree to which this happened in this sample did not reach statistical significance (P =.13 by Friedman test). By MFC, the median absolute change in disease burden after 1 dose of pembrolizumab relative to baseline was +0.022% (range, −1.1% to +1.5%). Of the 2 patients enrolled with only PCR-positive disease, 1 patient had an increase in disease burden such that it became detectable by MFC, and both patients had disease increase by PCR. Out of the 6 Ph+ patients, 5 had disease increase by PCR. Three patients had frank hematological relapse (≥5% blasts in the marrow) while receiving treatment with pembrolizumab. At data cutoff, 5 patients had died, all related to disease progression. Median overall survival was 12.7 months (range, 5.9-30.4 months), and median morphologic relapse-free survival was 3.1 months (range, 1.2-30.4 months; Figure 2).
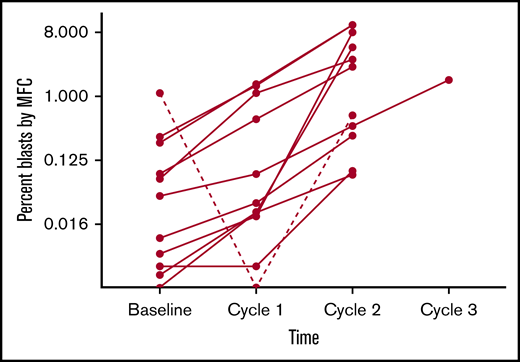
Changes in burden of ALL as determined by MFC of BMAs collected before and after each cycle of pembrolizumab. To better discriminate changes at the lower range of detection, blast percentage is plotted with a base-8 logarithmic scale. This graph depicts outcomes for the 11 patients who had disease detectable by MFC during study treatment. The course for the 1 patient who experienced a complete MRD response is depicted as a hashed line.
Changes in burden of ALL as determined by MFC of BMAs collected before and after each cycle of pembrolizumab. To better discriminate changes at the lower range of detection, blast percentage is plotted with a base-8 logarithmic scale. This graph depicts outcomes for the 11 patients who had disease detectable by MFC during study treatment. The course for the 1 patient who experienced a complete MRD response is depicted as a hashed line.
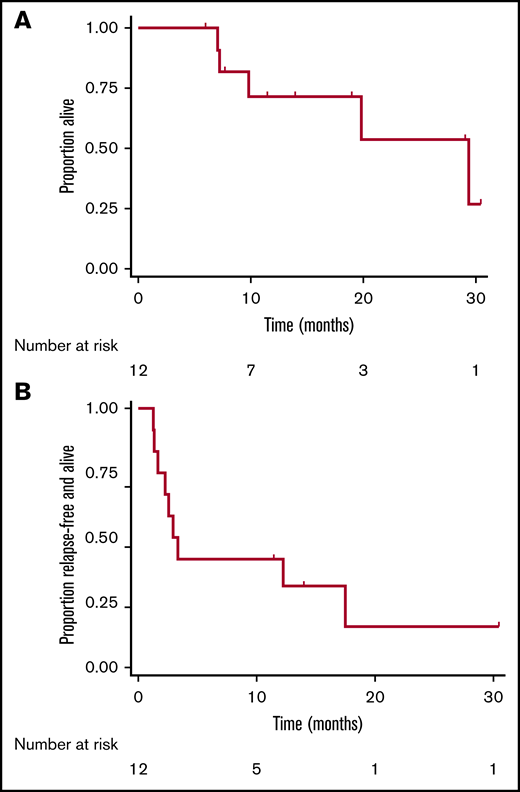
Time-to-event analyses following treatment with pembrolizumab. Kaplan-Meier curves depicting overall survival (A) and morphologic relapse-free survival (B) after administration of pembrolizumab in adults with ALL and MRD. Tick mark represent censored events.
Time-to-event analyses following treatment with pembrolizumab. Kaplan-Meier curves depicting overall survival (A) and morphologic relapse-free survival (B) after administration of pembrolizumab in adults with ALL and MRD. Tick mark represent censored events.
Because 1 of 12 patients did have a complete MRD response, the statistical design of the trial would have allowed enrollment of another 9 patients to more precisely estimate the response rate. However, since this single response was so transient, the principal investigator opted to terminate the study after completing the first stage of enrollment.
Five grade ≥3 AEs were deemed related to pembrolizumab: 3 incidences of grade 3 hypertension (13% of total cycles administered), 1 incidence of grade 3 Stevens-Johnson syndrome, and 1 incidence of grade 4 neutrophil count decreased. As alluded to above, the 1 patient who experienced a complete albeit transient MRD response was the only patient to experience an immune-related AE. One month after the second and final dose of pembrolizumab, the patient noted painful and pruritic papular lesions that were most prominent on the palms, soles, and distal legs, with some involvement on the anterior thighs and proximal arms and forearms. Many of these papules evolved toward frank bullae, especially at the acral extremities. A punch biopsy of one of these lesions on the right medial foot noted interface dermatitis by hematoxylin and eosin staining. A second biopsy of a lesion on the left lateral foot showed granular dermal-epidermal junction and superficial perivascular C3 deposition by direct immunofluorescence. There was no obvious evidence of mucosal or conjunctival involvement. After initiation of prednisone, all lesions resolved within days.
Correlative analyses of immunologic biomarkers
Levels of serum cytokines and inflammatory markers of interest were measured at enrollment and after cycle 2 of pembrolizumab (supplemental Figure 1). We also evaluated changes in the immunophenotypic profile of circulating leukocytes by flow cytometry at these same timepoints (supplemental Table 1). Due to the early termination of the trial and small sample size, limited analyses of these data were possible. That said, there was a statistically significant decrease in the serum levels of interleukin-15 and transforming growth factor β in patients who received 2 doses of pembrolizumab by 2-sided paired Wilcoxon tests. Lastly, among 2 of 3 patients who had previously received CD19 CAR-T cells and from whom such data were available, neither had evidence of persistent CAR-T cells in peripheral blood prior to enrollment (data not shown).
Discussion
Various new approaches to immunotherapy for cancer have evolved over the past 10 years. For B-cell malignancies, particularly B-cell ALL, the CD19-targeted therapies CAR-T cells and blinatumomab have demonstrated impressive short-term response rates and the potential for durable remissions in patients who historically would expect to have survival of <1 year with traditional cytotoxic chemotherapy.6,18,19 On the other hand, inhibition of PD-1/PD-L1 signaling has proven to be effective across a broad range of malignancies without restriction based on tissue of origin or immunophenotype. Unfortunately, our experience with pembrolizumab in this single-agent study in adults with ALL and MRD yielded minimal (but not zero) evidence of efficacy. These results suggest that this particular therapeutic approach is unlikely to have a role in the continuously-evolving landscape of treating this challenging disease. However, and despite our small study population, some of our observations could inform and justify further investigation of the use of these agents in more specific situations.
We observed only one transient response among 12 treated patients. While little can be concluded with confidence, a few things about this patient’s experience are noteworthy. First, this was 1 of 3 patients enrolled who had previously received other forms of adoptive immunotherapy; this patient underwent both haploidentical allogeneic HCT and CD19 CAR-T cell therapy years before receiving pembrolizumab on this trial. Upregulation of PD-L1 on malignant cells and increased markers of T-cell exhaustion (including PD-1) may represent mechanisms of resistance to HCT and CD19-directed immunotherapy.20,21 This may have rendered the patient’s disease more sensitive to PD-1 inhibition. This patient was also the only one to experience an immune-related AE. It is therefore tempting to speculate that the antileukemia effect observed might have been related to the immune response that led to the patient’s Stevens-Johnson syndrome. Lastly, for a study of patients exclusively with MRD, this patient’s disease burden was relatively high at enrollment. Though it is possible that the single timepoint at which a response was assessed (ie, after cycle 1) was due to sampling error from a bone marrow aspirate (BMA), the following details are noteworthy: relapse of this patient’s disease was first identified based on a peripheral blood PCR test ∼4 weeks before the first dose of pembrolizumab, during which interval no further therapy was administered. Thus, even if the BMA performed after cycle 1 of pembrolizumab was hemodiluted, the absence of detectable disease by both MFC and PCR suggests a reduction in overall disease burden.
Despite these interesting observations from the 1 responder, it is perhaps more instructive to consider reasons why pembrolizumab was ineffective in this patient population. Enrolling only patients with MRD should have avoided issues with excessive disease burden that could have deleteriously affected our results. As chemotherapy for ALL is particularly immunosuppressive (eg, corticosteroids, cyclophosphamide, and methotrexate), prior treatment may have left patients without an adequate immunologic reserve to respond to PD-1 inhibition. That said, this is seemingly not an issue with Hodgkin lymphoma, where similar drug classes are used prior to checkpoint inhibitors, as well as high-dose therapy and autologous stem cell transplantation. Further, our limited correlative analyses suggest some degree of immunologic effects of pembrolizumab in our patient population (supplemental Figure 1; supplemental Table 1). As for other possible explanations for inefficacy of pembrolizumab, tumor mutational burden has been correlated with response to anti–PD-1 antibodies.22 Even with its relatively low mutational rate, an analysis of somatic mutations in pediatric ALL predicted immunogenicity in vitro, suggesting such an approach could still be efficacious.23 Lastly, it may be that had we enrolled more patients with prior immunotherapy failures, we may have enriched for a population of patients more likely to respond to this approach. Despite our relatively discouraging results, we did not observe any cases of “hyperprogression” like that seen in adult T-cell leukemia/lymphoma.24
Notwithstanding our experience, there do appear to be viable paths forward to continue exploration of this highly impactful class of drugs in ALL. As noted above, upregulation of PD-1/PD-L1 signaling has been posited as a mechanism of resistance to CD19-directed immunotherapy, which very likely will stimulate efforts to add inhibitors of this pathway to improve outcomes with blinatumomab.20 Indeed, such trials combining blinatumomab with immune checkpoint inhibitors are already underway, early results from which have suggested safety and activity of these combinations.25,26 Similar studies have already begun using PD-1/PD-L1 inhibitors after CD19 CAR-T cells, one of which in pediatric ALL did demonstrate evidence of activity against bulky extramedullary disease.27-29 There are also other immune checkpoint inhibitors that may prove to yield more potent single-agent activity in ALL.30 Overall, based on the collective results to date, it would seem that checkpoint inhibitors have the most viable role in ALL as either a way to overcome resistance to or as an adjuvant for other more potent immunotherapeutic approaches. Our experience in this trial would argue against their use as a single agent following failure of cytotoxic chemotherapy.
Beyond the specific therapeutic strategy employed, our trial will hopefully advance the use of MRD as both a primary end point and an eligibility criterion for novel approaches. This is particularly true for a disease like ALL, where methods of MRD detection and its importance are well established. Patients with MRD represent a high-risk population whose disease burden is sufficiently high to quantitatively evaluate efficacy but low enough to not pose patients an imminent threat if the proposed intervention is ineffective. As MRD detection becomes more robust in other diseases, designing trials specifically for this patient population could expand access to novel agents and accelerate our approach to evaluating them.
In conclusion, while single-agent pembrolizumab in relatively unselected adults with ALL and MRD was relatively well tolerated, we saw minimal evidence of efficacy. This approach is unlikely to be a viable therapeutic method moving forward. However, as more is understood regarding mechanisms of resistance and failure of other more established forms of immunotherapy, inhibition of PD-1/PD-L1 signaling may prove to be a useful adjunct or rescue strategy. These data provide an important baseline experience onto which this approach may be developed.
Due to regulatory and contractual requirements, individual participant data will not be shared. Queries regarding the clinical protocol should be directed to the corresponding author, Ryan D. Cassaday (e-mail: cassaday@uw.edu).
Acknowledgments
The authors would like to thank Margaret Nartea and staff at both the University of Washington Hematopathology Laboratory and Fred Hutchinson Cancer Research Center Immune Monitoring Core for technical assistance with our correlative analyses.
This study was funded by the Merck Investigator Sponsor Program (MISP #52421) and donations from the family of Ann Molitor and Zuri Hector.
Authorship
Contribution: R.D.C. and E.H.E. contributed to study design; R.D.C., K.-L.A.G., J.R.F., and P.T.N. collected data; R.D.C., J.R.F., M.-E.M.P., and C.J.T. analyzed data; P.A.S. performed statistical analyses; R.D.C. and K.-L.A.G. wrote the manuscript; and all authors edited and reviewed drafts and approved submission of the final version of the manuscript.
Conflict-of-interest disclosure: R.D.C. has received research funding from Amgen, Kite/Gilead, Merck, Pfizer, and Vanda Pharmaceuticals and honoraria/consulting from Amgen and Pfizer, and his spouse is employed by and owns stock in Seattle Genetics. J.R.F. has received research funding from Merck. M.-E.M.P. has received research funding from Pfizer, Biosight, Nohla, Trillium, and FLX Bio as well as honoraria from Genentech. C.J.T. receives research funding from Juno Therapeutics, Nektar Therapeutics, TCR2 Therapeutics, and AstraZeneca; is a member of scientific advisory boards and has options in Precision Biosciences, Eureka Therapeutics, Caribou Biosciences, Myeloid Therapeutics, and ArsenalBio; serves on scientific advisory boards for T-CURX and Century Therapeutics; has served on advisory boards for Nektar Therapeutics, Allogene, Kite/Gilead, Novartis, Humanigen, PACT Pharma, and AstraZeneca; and has patents licensed to Juno Therapeutics. P.T.N. has served on advisory boards for Pfizer and EMD Serono, and his institution receives research support from Bristol-Myers Squibb and EMD Serono. The remaining authors declare no competing financial interests.
Correspondence: Ryan D. Cassaday, Seattle Cancer Care Alliance, 825 Eastlake Ave E, Mailstop CE3-300, Seattle, WA 98136; e-mail: cassaday@uw.edu.

