

Key Points
Sonidegib 400 mg once daily + ruxolitinib 20 mg twice daily was the RP2D for JAK inhibitor–naive patients with MF.
The overall benefit of the combination was relatively modest compared with ruxolitinib monotherapy.

Abstract
The sonidegib and ruxolitinib combination was assessed in an open-label study in JAK inhibitor-naive patients with myelofibrosis (MF). The primary objective of phase 1b was to establish the maximum tolerated dose (MTD) and/or recommended phase 2 dose (RP2D) and phase 2 was to assess spleen volume reduction at weeks 24 and 48. Fifty patients were enrolled. In the dose-escalation phase (n = 23), doses for sonidegib once daily/ruxolitinib twice daily were 400/10 mg (level 1, n = 8), 400/15 mg (level 2, n = 10), and 400/20 mg (level 3, n = 5). Two patients had dose-limiting toxicity at level 2: increased blood creatine phosphokinase (grades 3 and 4, n = 1 each). MTD/RP2D was determined as sonidegib 400 mg daily + ruxolitinib 20 mg twice daily. In phase 1b expansion and phase 2 stage 1 (n = 27), by weeks 24 and 48, ≥35% reduction in spleen volume was observed in 44.4% and 29.6% patients, respectively. By weeks 24 and 48, 42.0% and 26.0% patients had ≥50% reduction in Myelofibrosis Symptom Assessment Form total symptom score, respectively. Most common treatment-related adverse events (grade 3/4) were increased blood creatine phosphokinase (18%), anemia (14%), and thrombocytopenia (12%). Four deaths were reported due to multiple organ dysfunction syndrome (on-treatment; no relationship with study treatment), acute myeloid leukemia, MF progression, and aspiration pneumonia. Although well tolerated, this combination will not be further developed in MF patients due to modest overall benefit compared with historical ruxolitinib monotherapy. This trial was registered at www.clinicaltrials.gov as #NCT01787552.
Introduction
Myelofibrosis (MF), whether primary myelofibrosis (PMF) or following polycythemia vera (PV) or following essential thrombocythemia (ET), is a myeloproliferative neoplasm (MPN) characterized by cytopenias, extramedullary hematopoiesis, megakaryocytic dysplasia, bone marrow (BM) fibrosis, and/or systemic symptoms occurring due to elevated levels of inflammatory and proangiogenic cytokines.1,2 Most patients with MF harbor a mutation in the gene encoding JAK2, MPL, or CALR, causing dysregulation of the Janus kinase (JAK)/signal transducer and activator of transcription pathway, which plays a central role in the pathogenesis of the disease.1
Ruxolitinib is a first-in-class JAK1/2 inhibitor (JAKi) approved in 2011 by the US Food and Drug Administration for patients with intermediate- or high-risk MF and in patients with PV who have had an inadequate response to or are intolerant of hydroxyurea.3 The approval of ruxolitinib in MF was based on 2 randomized phase 3 clinical trials (COMFORT-I and COMFORT-II). In both of these studies, ruxolitinib demonstrated marked and sustained clinical benefits in terms of reduction in spleen size and improvements in symptomatic burden. It was generally well tolerated, and the adverse events (AEs) were usually managed with dose modifications.1,4 Despite clear benefits, ruxolitinib therapy is noncurative and is associated with treatment-related anemia, which may exacerbate preexisting MF-related anemia. Novel strategies are required to improve the anticlonal activity and response of ruxolitinib.5
The hedgehog (Hh) signaling pathway has been implicated in the pathogenesis of several hematologic malignancies, including MF, and plays a role in proliferation, differentiation, and survival during embryonic development and stem cell maintenance in adults.5,6 Activated Hh signaling can promote carcinogenesis and maintenance of leukemic stem cells. In the resting state, the transmembrane protein “Patched” (PTCH) inhibits the activity of a membrane spanning receptor called “Smoothened” (SMO). Upon Hh ligand binding, activated SMO signals through a complex of cystolic proteins, resulting in the activation of Glioma-associated oncogene homolog (Gli) transcription factors and their subsequent nuclear translocation to induce Hh target genes, such as Gli 1, PTCH1, cyclin D1, bcl-2, N-myc, and secreted frizzled-related protein 1. The Gli transcription factors, the downstream effectors of Hh signaling,7 promote cell proliferation, differentiation, and survival.8 Therefore, the expression of Gli1 messenger RNA in tumor or relevant surrogate tissues constitute a reliable indicator of Hh pathway activity.9 Several Hh pathway inhibitors are in clinical development for myeloid malignancies.6,10,11 One such Hh inhibitor, glasdegib, has recently been approved by the US Food and Drug Administration for the treatment of acute myeloid leukemia (AML) in combination with low-dose cytarabine.12 Sonidegib, an orally bioavailable, small molecule Hh inhibitor that targets the membrane protein, SMO, has been approved in Switzerland for the treatment of adult patients with advanced basal cell carcinoma (BCC) and in the United States and European Union for the treatment of adult patients with locally advanced BCC that has recurred following surgery or radiation therapy, or those who are not candidates for surgery or radiation therapy.13
A preclinical study demonstrated 100-fold overexpression of Gli and PTCH1 in granulocytes from patients with MPNs, as well as upregulation of the Hh pathway in murine MPN models. This study also demonstrated a synergistic effect of combination therapy of sonidegib and ruxolitinib resulting in significant reduction in spleen weight, mutant JAK2 allele burden, and BM fibrosis compared with either drug alone.14 Potential antileukemic stem cell activity and synergistic action with ruxolitinib provided the rationale for investigating this combination therapy. This phase 1b/phase 2 study aimed to assess the safety and efficacy of the oral combination of sonidegib and ruxolitinib in patients with MF.
Methods
Patient population
Adult patients with PMF, post-PV MF, or post-ET MF without prior therapy with JAKi were included in the study. Patients had palpable splenomegaly and were classified as intermediate risk level 1, intermediate risk level 2, or high risk as defined by the International Working Group.15 Further details on the eligibility criteria are provided in the supplemental Material. The study protocol and all amendments were reviewed by the Independent Ethics Committee, the Institutional Review Board, or the Research Ethics Board for each center. The study was conducted according to the ethical principles of the Declaration of Helsinki. All patients provided written informed consent.
Study design
This study was an open-label, multicenter, dose-finding, phase 1b/2 study. In the phase 1b part of the study, there was a dose-escalation/safety expansion part to determine the maximum tolerated dose (MTD) and/or identify the recommended phase 2 dose (RP2D) and evaluate the safety for the combination of sonidegib once daily and ruxolitinib twice daily in patients with PMF, post-PV MF, or post-ET MF. The phase 2 part was aimed to assess the efficacy of the drug combination on spleen volume reduction (supplemental Figure 1).
Phase 1b (dose-escalation and safety expansion).
This phase was designed in 2 parts. In the dose-escalation part, patient cohorts received increasing doses of sonidegib and ruxolitinib until an MTD/RP2D was established. For each drug in the combination, the starting dose was the one that demonstrated clinical activity while being safe (at or near 50% of their respective MTDs). Hence, the starting dose of the combination was selected as sonidegib 400 mg daily and ruxolitinib 10 mg twice daily. In the dose expansion part, confirmatory patients were enrolled at each MTD/RP2D. Approximately 36 patients were planned to be treated in the dose-escalation and safety expansion part.
Phase 2.
In this phase, patients received the dose(s) identified as the MTD/RP2D. Approximately 46 patients were planned to be enrolled in 2 stages in phase 2. However, stage 2 of phase 2 was not conducted due to limited activity observed with the combination therapy in the interim analysis of stage 1 of phase 2.
All patients remained on study treatment for at least 2 years after the first dose or until they permanently discontinued both the study drugs due to any cause. A safety follow-up was conducted 30 days (+3 days) after the last dose of study medication.
Objectives and endpoints
Phase 1b.
The primary objective was to establish the MTD and/or RP2D of the coadministration of sonidegib and ruxolitinib in patients with MF who have not previously received JAKi therapy. The primary endpoint was the incidence of dose-limiting toxicities (DLTs) in the first 6 weeks of treatment.
The key secondary objectives were to evaluate the safety of the coadministration of sonidegib and ruxolitinib in patients with MF and to characterize the single- and multiple-dose pharmacokinetics (PK) following the coadministration of sonidegib and ruxolitinib.
Phase 2.
The primary objective was to assess the efficacy of the coadministration of sonidegib and ruxolitinib on spleen volume reduction as determined by the proportion of patients achieving ≥35% reduction in spleen volume from baseline by a centrally reviewed magnetic resonance imaging (MRI)/computed tomography (CT) scan at the end of week 24 and week 48.
The key secondary objectives were to assess the effect of the combination on BM fibrosis and biomarkers as a function of the molecular disease characterization of MF, safety, PK, and the effect on MF-associated symptom burden.
Assessments
A Bayesian Logistic Regression Model using the escalation with overdose control principle was used for dose level selection and determination of the MTD and/or RP2D.
An MRI/CT scan of the abdomen was performed for patients enrolled to the confirmatory MTD level(s) in the phase 1b part and for all patients enrolled in the phase 2 part of the study. The response assessment in the phase 1b part of the study consisted of comparing the spleen length (measured by manual palpation) at the end of week 24 with week 1, day 1 measurement.
AEs and serious AEs (SAEs) were evaluated as part of safety assessments.
Patient-reported outcomes were assessed using the 7-day modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.016 and the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire. JAK2 mutation status, V617F allele burden, and Gli1 expression were also measured. CALR mutations were not assessed in this study as these were described after the initiation of the trial.
Serial blood samples for PK evaluation were collected within the 24 hours after drug administration on week 1, day 1 and week 9, day 1. Plasma concentrations of sonidegib and ruxolitinib were determined by a validated liquid chromatography tandem mass spectrometry. PK parameters for sonidegib and ruxolitinib were derived by standard noncompartmental analysis methods by using WinNonlin software version 6.4 (Pharsight, Mountain View, CA).
Statistical analyses
Phase 1b.
Following the principle of escalation with overdose control, after each cohort of patients, the recommended dose combination was the one with the highest posterior probability of DLT in the target interval (16%, 35%) among the doses fulfilling the overdose-control criterion of <25% (posterior probability) chance of excessive toxicity. A clinical synthesis of the available toxicity information, PK, pharmacodynamic, and efficacy information as well as the recommendations from the Bayesian model was used to determine the combination dose for the next cohort.
Phase 2.
Patients were considered as responders if they achieved >35% reduction in the spleen volume from baseline to week 24 or week 48, provided the patient had evaluable baseline and week 24 or week 48 spleen volume assessments, and the patient did not experience a protocol-defined progression event prior to the week 24 or week 48 visit, and the calculated percentage change was ≤−35%. The uniformly minimum-variance unbiased estimator, in patients achieving 35% spleen volume reduction from baseline as determined by an MRI/CT scan, has been estimated in phase 2with the exact 2-sided 95% confidence interval (CI).
The patients were categorized into various analysis sets, the details of which are provided in supplemental Table 1.
Results
Patients
Fifty patients were enrolled in the study (phase 1b dose escalation, n = 23; phase 1b dose expansion and phase 2 stage 1, n = 27). The median age (range) of patients was 68.5 (42 to 83) years and 70% were male. Overall, 52% of the patients had PMF, and 82% had grade 2 or 3 BM fibrosis. The baseline characteristics of patients are summarized in Table 1.
Baseline patient characteristics
| Demographic variable/disease history | Phase 1b dose-escalation phase | Phase 1b dose expansion phase and phase 2 stage 1 | |||
|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | All patients (N = 50) | |
| Age, median (range), y | 73.0 (63-77) | 58.5 (42-75) | 71.0 (42-74) | 69.0 (44-83) | 68.5 (42-83) |
| Age ≥60 y, n (%) | 8 (100) | 5 (50.0) | 4 (80.0) | 21 (77.8) | 38 (76.0) |
| Male, n (%) | 7 (87.5) | 5 (50.0) | 4 (80.0) | 19 (70.4) | 35 (70.0) |
| ECOG PS, n (%) | |||||
| 0 | 1 (12.5) | 2 (20.0) | 3 (60.0) | 13 (48.1) | 19 (38.0) |
| 1 | 6 (75.0) | 8 (80.0) | 2 (40.0) | 14 (51.9) | 30 (60.0) |
| 2 | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.0) |
| Diagnosis of disease, n (%) | |||||
| PMF | 1 (12.5) | 7 (70.0) | 2 (40.0) | 16 (59.3) | 26 (52.0) |
| Post-PV MF | 5 (62.5) | 2 (20.0) | 1 (20.0) | 4 (14.8) | 12 (24.0) |
| Post-ET MF | 2 (25.0) | 1 (10.0) | 2 (40.0) | 7 (25.9) | 12 (24.0) |
| Spleen parameters | |||||
| Spleen size measured by palpation, mean ± SD, cm | 17.4 (8.78) | 13.1 (10.14) | 15.5 (5.02) | 11.6 (7.01) | 13.2 (7.93) |
| Spleen volume measured by MRI/CT scan, mean ± SD, cm3 | — | — | — | 1953.6 (932.03) | — |
| BM fibrosis grade (most recent diagnosis result prior to study entry), n (%) | |||||
| 0 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (7.4) | 2 (4.0) |
| 1 | 0 (0.0) | 1 (10.0) | 0 (0.0) | 3 (11.1) | |
| 2 | 5 (62.5) | 5 (50.0) | 2 (40.0) | 9 (33.3) | 21 (42.0) |
| 3 | 3 (37.5) | 3 (30.0) | 3 (60.0) | 11 (40.7) | 20 (40.0) |
| Missing | 0 (0.0) | 1 (10.0) | 0 (0.0) | 2 (7.4) | 3 (6.0) |
| IPSS risk factor at time of study entry, n (%) | |||||
| Low | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Intermediate-1 | 1 (12.5) | 5 (50.0) | 0 (0.0) | 5 (18.5) | 11 (22.0) |
| Intermediate-2 | 1 (12.5) | 2 (20.0) | 3 (60.0) | 4 (14.8) | 10 (20.0) |
| High | 6 (75.0) | 3 (30.0) | 2 (40.0) | 18 (66.7) | 29 (58.0) |
| JAK2 V617F allele burden,*n (%) | |||||
| Positive | 8 (100.0) | 8 (80.0) | 5 (100.0) | 23 (85.2) | 44 (88) |
| Negative | 0 (0.0) | 2 (20.0) | 0 (0.0) | 4 (14.8) | 6 (12) |
| Demographic variable/disease history | Phase 1b dose-escalation phase | Phase 1b dose expansion phase and phase 2 stage 1 | |||
|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | All patients (N = 50) | |
| Age, median (range), y | 73.0 (63-77) | 58.5 (42-75) | 71.0 (42-74) | 69.0 (44-83) | 68.5 (42-83) |
| Age ≥60 y, n (%) | 8 (100) | 5 (50.0) | 4 (80.0) | 21 (77.8) | 38 (76.0) |
| Male, n (%) | 7 (87.5) | 5 (50.0) | 4 (80.0) | 19 (70.4) | 35 (70.0) |
| ECOG PS, n (%) | |||||
| 0 | 1 (12.5) | 2 (20.0) | 3 (60.0) | 13 (48.1) | 19 (38.0) |
| 1 | 6 (75.0) | 8 (80.0) | 2 (40.0) | 14 (51.9) | 30 (60.0) |
| 2 | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.0) |
| Diagnosis of disease, n (%) | |||||
| PMF | 1 (12.5) | 7 (70.0) | 2 (40.0) | 16 (59.3) | 26 (52.0) |
| Post-PV MF | 5 (62.5) | 2 (20.0) | 1 (20.0) | 4 (14.8) | 12 (24.0) |
| Post-ET MF | 2 (25.0) | 1 (10.0) | 2 (40.0) | 7 (25.9) | 12 (24.0) |
| Spleen parameters | |||||
| Spleen size measured by palpation, mean ± SD, cm | 17.4 (8.78) | 13.1 (10.14) | 15.5 (5.02) | 11.6 (7.01) | 13.2 (7.93) |
| Spleen volume measured by MRI/CT scan, mean ± SD, cm3 | — | — | — | 1953.6 (932.03) | — |
| BM fibrosis grade (most recent diagnosis result prior to study entry), n (%) | |||||
| 0 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (7.4) | 2 (4.0) |
| 1 | 0 (0.0) | 1 (10.0) | 0 (0.0) | 3 (11.1) | |
| 2 | 5 (62.5) | 5 (50.0) | 2 (40.0) | 9 (33.3) | 21 (42.0) |
| 3 | 3 (37.5) | 3 (30.0) | 3 (60.0) | 11 (40.7) | 20 (40.0) |
| Missing | 0 (0.0) | 1 (10.0) | 0 (0.0) | 2 (7.4) | 3 (6.0) |
| IPSS risk factor at time of study entry, n (%) | |||||
| Low | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Intermediate-1 | 1 (12.5) | 5 (50.0) | 0 (0.0) | 5 (18.5) | 11 (22.0) |
| Intermediate-2 | 1 (12.5) | 2 (20.0) | 3 (60.0) | 4 (14.8) | 10 (20.0) |
| High | 6 (75.0) | 3 (30.0) | 2 (40.0) | 18 (66.7) | 29 (58.0) |
| JAK2 V617F allele burden,*n (%) | |||||
| Positive | 8 (100.0) | 8 (80.0) | 5 (100.0) | 23 (85.2) | 44 (88) |
| Negative | 0 (0.0) | 2 (20.0) | 0 (0.0) | 4 (14.8) | 6 (12) |
—, not available; ECOG PS, Eastern Cooperative Oncology Group Performance Status; IPSS, International Prognostic Scoring System; LLOQ, lower limit of quantitation; SD, standard deviation.
The interpretation of mutation is based on the mean allele burden value compared with LLOQ, which is equal to 5.0 in this study. If ≥LLOQ, then it is positive. Any value <LLOQ will be considered as mutation negative.
At the time of early study termination, all 50 patients had discontinued the study. The primary reasons for discontinuation included AEs (50%), physician decision (14%), disease progression (12%), and patient/guardian decision (12%) (Table 2).
Patient disposition by study phase and treatment in phase 1b and phase 2 stage 1 (full analysis set)
| Phase 1b dose-escalation phase, n (%) | Phase 1b dose expansion phase and phase 2 stage 1, n (%) | ||||
|---|---|---|---|---|---|
| Reasons for discontinuation of treatment | Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | All patients (N = 50), n (%) |
| AEs* | 2 (25.0) | 5 (50.0) | 2 (40.0) | 16 (59.3) | 25 (50.0) |
| Completed | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 1 (2.0) |
| Death† | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (3.7) | 1 (2.0) |
| Physician’s decision | 2 (25.0) | 2 (20.0) | 1 (20.0) | 2 (7.4) | 7 (14.0) |
| Progressive disease | 2 (25.0) | 2 (20.0) | 1 (20.0) | 1 (3.7) | 6 (12.0) |
| Study terminated by sponsor | 0 (0.0) | 0 (0.0) | 1 (20.0) | 3 (11.1) | 4 (8.0) |
| Patient/guardian decision | 2 (25.0) | 0 (0.0) | 0 (0.0) | 4 (14.8) | 6 (12.0) |
| Phase 1b dose-escalation phase, n (%) | Phase 1b dose expansion phase and phase 2 stage 1, n (%) | ||||
|---|---|---|---|---|---|
| Reasons for discontinuation of treatment | Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | All patients (N = 50), n (%) |
| AEs* | 2 (25.0) | 5 (50.0) | 2 (40.0) | 16 (59.3) | 25 (50.0) |
| Completed | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 1 (2.0) |
| Death† | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (3.7) | 1 (2.0) |
| Physician’s decision | 2 (25.0) | 2 (20.0) | 1 (20.0) | 2 (7.4) | 7 (14.0) |
| Progressive disease | 2 (25.0) | 2 (20.0) | 1 (20.0) | 1 (3.7) | 6 (12.0) |
| Study terminated by sponsor | 0 (0.0) | 0 (0.0) | 1 (20.0) | 3 (11.1) | 4 (8.0) |
| Patient/guardian decision | 2 (25.0) | 0 (0.0) | 0 (0.0) | 4 (14.8) | 6 (12.0) |
The most frequently reported (≥5% of patients) AEs leading to study drug discontinuation were increased blood creatine phosphokinase, thrombocytopenia, muscle spasms, AML, and alopecia.
On-treatment death due to multiple organ dysfunction syndrome: 4 deaths were reported during the study; the remaining 3 patients discontinued their treatment due to “progressive disease” (n = 2) and an AE (n = 1).
Dose and duration of exposure
In the dose-escalation phase, dose levels included for the sonidegib daily/ruxolitinib twice daily combination were 400 mg/10 mg (dose level 1, n = 8), 400 mg/15 mg (dose level 2, n = 10), and 400 mg/20 mg (dose level 3, n = 5). The median duration of exposure to ruxolitinib was 424.0 days (30.0 to 1505.0 days) and to sonidegib was 411.5 days (30.0 to 1505.0). Overall, 88.0% (n = 44) of patients had at least 1 dose reduction/interruption; 70% of patients had at least 1 dose reduction, and 60% of patients had at least 1 dose interruption (supplemental Table 2).
MTD/RP2D
The dose-determining set comprised 29 patients. No DLTs were observed at the first (n = 8) and third (n = 5) dose levels. Two patients experienced increased blood creatine phosphokinase as a DLT (1 patient each for grade 3 and grade 4) at the second dose level (n = 10). Both events were suspected to be study drug related and resulted in temporary dose interruption (Table 3).
DLTs in phase 1b (safety analysis set)
| Investigation | Phase 1b dose-escalation phase, n (%) | Phase 1b dose expansion phase, n (%) | |||
|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | All patients (N = 50), n (%) | |
| Increased blood creatine phosphokinase | 0 (0.0) | 2 (20.0) | 0 (0.0) | 1 (3.7) | 3 (6.0) |
| Investigation | Phase 1b dose-escalation phase, n (%) | Phase 1b dose expansion phase, n (%) | |||
|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | All patients (N = 50), n (%) | |
| Increased blood creatine phosphokinase | 0 (0.0) | 2 (20.0) | 0 (0.0) | 1 (3.7) | 3 (6.0) |
The MTD and RP2D were determined to be sonidegib 400 mg daily + ruxolitinib 20 mg twice daily. In the phase 1b dose expansion phase, 7 patients were enrolled: 6 were included in the dose-determining set and 1 had increased blood creatine phosphokinase (grade 4) as a DLT. Sonidegib 400 mg daily + ruxolitinib 20 mg twice daily was further confirmed as the MTD/RP2D in the expansion cohort.
Efficacy
Of the 27 patients in phase 1b expansion and phase 2 stage 1, a ≥35% reduction in spleen volume (per MRI/CT scan) was observed in 44.4% (95% CI: 25.48 to 64.67) of patients by week 24 and in 29.6% (95% CI: 13.75 to 50.18) of patients by week 48. In phase 1b expansion and phase 2 stage 1 (n = 27), 24 (88.9%) patients demonstrated reduction in spleen volume from baseline (Figure 1). At least 50% reduction in spleen size by manual palpation was observed in 29.6% (95% CI: 13.75 to 50.18) by week 24 and in 11.1% (95% CI: 2.35 to 29.16) by week 48.
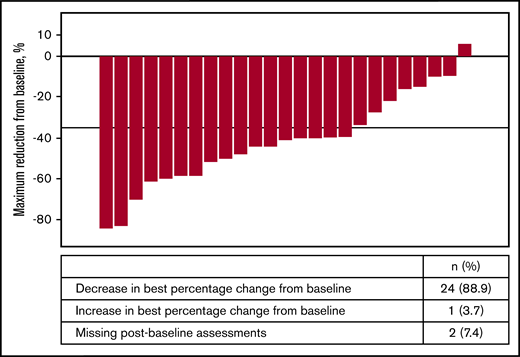
Waterfall plot of best percentage reduction in spleen volume (cm3) as per MRI/CT for phase 1b expansion phase and phase 2 stage 1 (full analysis set).
Waterfall plot of best percentage reduction in spleen volume (cm3) as per MRI/CT for phase 1b expansion phase and phase 2 stage 1 (full analysis set).
Across all patients (n = 50), at least 50% reduction in the MFSAF total symptom score (TSS) was observed in 42.0% of patients (n = 21) by week 24 and in 26.0% of patients (n = 13) by week 48. In phase 1b expansion and phase 2 stage 1 (n = 27), the mean change (standard deviation [SD]) in TSS was −6.5 (9.26) at week 24 (n = 19) and −8.0 (14.61) at week 48 (n = 10). This change was not significant. The best percentage change from baseline score for MFSAF TSS in phase 1b expansion and phase 2 stage 1 is shown in supplemental Figure 2.
The European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-C30 demonstrated no significant change in the functional, global health status/quality of life, and symptom scales.
Biomarkers and BM fibrosis
At baseline, 44 (88%) patients were JAK2 positive, and 6 (12%) patients were negative. In JAK2 mutated patients, the mean percentage change from baseline for JAK2 V617F allele burden ranged from −0.8% to −7.5% at the end of the treatment.
Overall, for Gli1 expression, there was a minimal mean fold-change at week 105 (end of treatment) from baseline, with values ranging from −0.2 to −1.4 (Table 4).
Gli1 expression by reverse transcription polymerase chain reaction (ΔCT) in whole blood by treatment in phase 1b and phase 2 stage 1
| Level | Phase 1b dose-escalation phase | Phase 1b dose expansion phase | All patients (N = 50) | ||
|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | ||
| Baseline | |||||
| n (%) | 8 (100) | 10 (100) | 5 (100) | 27 (100) | 50 (100) |
| Mean | 6.9 | 6.5 | 6.6 | 6.4 | 6.52 |
| SD (CV%) | 0.80 (11.5) | 0.90 (13.8) | 0.45 (6.8) | 0.93 (14.5) | 0.79 (13.1) |
| Week 25, day 1 (fold-change from baseline) | |||||
| n (%) | 6 (75.0) | 7 (70.0) | 5 (100) | 22 (81.5) | 40 (80) |
| Mean | −0.7 | −0.8 | −1.4 | −0.9 | −0.92 |
| SD (CV%) | 1.41 (−206.4) | 1.55 (−199.8) | 1.77 (−124.7) | 2.08 (−229.6) | 1.68 (−207.8) |
| Week 49, day 1 (fold-change from baseline) | |||||
| n (%) | 4 (50.0) | 3 (30.0) | 4 (80.0) | 14 (51.9) | 25 (50) |
| Mean | −1.0 | −0.2 | −1.3 | −1.7 | −1.34 |
| SD (CV%) | 1.56 (−160.6) | 1.45 (−706.0) | 2.04 (−155.8) | 2.45 (−147.0) | 1.82 (−217.7) |
| End of treatment (fold-change from baseline) | |||||
| n (%) | 4 (50.0) | 8 (80.0) | 1 (20.0) | 22 (81.5) | 35 (70) |
| Mean | −1.0 | −0.2 | −1.0 | −1.4 | −1.07 |
| SD (CV%) | 1.57 (−155.7) | 1.76 (−871.6) | 0 (0.0) | 2.15 (−156.6) | 1.78 (−315.5) |
| Level | Phase 1b dose-escalation phase | Phase 1b dose expansion phase | All patients (N = 50) | ||
|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | ||
| Baseline | |||||
| n (%) | 8 (100) | 10 (100) | 5 (100) | 27 (100) | 50 (100) |
| Mean | 6.9 | 6.5 | 6.6 | 6.4 | 6.52 |
| SD (CV%) | 0.80 (11.5) | 0.90 (13.8) | 0.45 (6.8) | 0.93 (14.5) | 0.79 (13.1) |
| Week 25, day 1 (fold-change from baseline) | |||||
| n (%) | 6 (75.0) | 7 (70.0) | 5 (100) | 22 (81.5) | 40 (80) |
| Mean | −0.7 | −0.8 | −1.4 | −0.9 | −0.92 |
| SD (CV%) | 1.41 (−206.4) | 1.55 (−199.8) | 1.77 (−124.7) | 2.08 (−229.6) | 1.68 (−207.8) |
| Week 49, day 1 (fold-change from baseline) | |||||
| n (%) | 4 (50.0) | 3 (30.0) | 4 (80.0) | 14 (51.9) | 25 (50) |
| Mean | −1.0 | −0.2 | −1.3 | −1.7 | −1.34 |
| SD (CV%) | 1.56 (−160.6) | 1.45 (−706.0) | 2.04 (−155.8) | 2.45 (−147.0) | 1.82 (−217.7) |
| End of treatment (fold-change from baseline) | |||||
| n (%) | 4 (50.0) | 8 (80.0) | 1 (20.0) | 22 (81.5) | 35 (70) |
| Mean | −1.0 | −0.2 | −1.0 | −1.4 | −1.07 |
| SD (CV%) | 1.57 (−155.7) | 1.76 (−871.6) | 0 (0.0) | 2.15 (−156.6) | 1.78 (−315.5) |
CV% means are derived as “SD/mean*100.” ΔCT, a target gene, is the difference between the target gene CT and the average of the control genes CTs. Fold-change is defined as follows: let Diff = (ΔCT)post − (ΔCT)pre; fold-change = 2^(Diff); if Diff ≥0; fold-change = −1*{2^(−1*Diff) }; if Diff <0.
CT, cycle threshold (raw CT results normalized).
BM biopsies were available in 41 (82%), 30 (60%), and 15 (30%) patients at baseline, week 25, and week 49, respectively. There was no significant improvement in the grade of fibrosis at week 25 and week 49 (supplemental Table 3).
Safety
All patients experienced at least 1 AE; 78% experienced grade 3 or 4 AEs. The most frequently reported AEs (≥20% of patients, all grades) are shown in Table 5. The most commonly reported AEs (≥10% of patients, grade 3/4) included anemia (40%), increased blood creatine phosphokinase (18%), and thrombocytopenia (14%).
AEs (≥20% of patients), regardless of study drug relationship (safety analysis set)
| Phase 1b dose-escalation phase, n (%) | Phase 1b dose expansion phase and phase 2 stage 1. n (%) | All patients (N = 50), n (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | |||||||
| All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | |
| Total | 8 (100) | 8 (100) | 10 (100) | 8 (80) | 5 (100) | 4 (80) | 27 (100) | 19 (70.4) | 50 (100) | 39 (78) |
| Muscle spasms* | 3 (37.5) | 0 (0.0) | 6 (60.0) | 1 (10.0) | 4 (80.0) | 0 (0.0) | 18 (66.7) | 1 (3.7) | 31 (62.0) | 2 (4.0) |
| Myalgia* | 1 (12.5) | 0 (0.0) | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 10 (37.0) | 2 (7.4) | 14 (28.0) | 2 (4.0) |
| Fatigue | 6 (75.0) | 1 (12.5) | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 8 (29.6) | 0 (0.0) | 17 (34.0) | 1 (2.0) |
| Pyrexia | 2 (25.0) | 2 (25.0) | 2 (20.0) | 0 (0.0) | 2 (40.0) | 0 (0.0) | 9 (33.3) | 2 (7.4) | 15 (30.0) | 4 (8.0) |
| Cough | 1 (12.5) | 0 (0.0) | 3 (30.0) | 0 (0.0) | 2 (40.0) | 0 (0.0) | 5 (18.5) | 0 (0.0) | 11 (22.0) | 0 (0.0) |
| Anemia | 5 (62.5) | 5 (62.5) | 4 (40.0) | 2 (20.0) | 4 (80.0) | 2 (40.0) | 17 (63.0) | 11 (40.7) | 30 (60.0) | 20 (40.0) |
| Thrombocytopenia | 3 (37.5) | 1 (12.5) | 2 (20.0) | 0 (0.0) | 3 (60.0) | 2 (40.0) | 7 (25.9) | 4 (14.8) | 15 (30.0) | 7 (14.0) |
| Diarrhea | 3 (37.5) | 0 (0.0) | 3 (30.0) | 0 (0.0) | 4 (80.0) | 0 (0.0) | 12 (44.4) | 3 (11.1) | 22 (44.0) | 3 (6.0) |
| Constipation | 2 (25.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 1 (20.0) | 0 (0.0) | 7 (25.9) | 0 (0.0) | 11 (22.0) | 0 (0.0) |
| Nausea | 3 (37.5) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 8 (29.6) | 0 (0.0) | 12 (24.0) | 0 (0.0) |
| Vomiting | 3 (37.5) | 0 (0.0) | 2 (20.0) | 0 (0.0) | 1 (20.0) | 0 (0.0) | 6 (22.2) | 1 (3.7) | 12 (24.0) | 1 (2.0) |
| Blood creatine phosphokinase increased* | 1 (12.5) | 0 (0.0) | 2 (20.0) | 2 (20.0) | 3 (60.0) | 1 (20.0) | 12 (44.4) | 6 (22.2) | 18 (36.0) | 9 (18.0) |
| Dysgeusia* | 3 (37.5) | 0 (0.0) | 4 (40.0) | 0 (0.0) | 3 (60.0) | 0 (0.0) | 9 (33.3) | 0 (0.0) | 19 (38.0) | 0 (0.0) |
| Alopecia* | 3 (37.5) | 0 (0.0) | 7 (70.0) | 0 (0.0) | 3 (60.0) | 0 (0.0) | 12 (44.4) | 0 (0.0) | 25 (50.0) | 0 (0.0) |
| Phase 1b dose-escalation phase, n (%) | Phase 1b dose expansion phase and phase 2 stage 1. n (%) | All patients (N = 50), n (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sonidegib 400 mg + ruxolitinib 10 mg (n = 8) | Sonidegib 400 mg + ruxolitinib 15 mg (n = 10) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 5) | Sonidegib 400 mg + ruxolitinib 20 mg (n = 27) | |||||||
| All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | |
| Total | 8 (100) | 8 (100) | 10 (100) | 8 (80) | 5 (100) | 4 (80) | 27 (100) | 19 (70.4) | 50 (100) | 39 (78) |
| Muscle spasms* | 3 (37.5) | 0 (0.0) | 6 (60.0) | 1 (10.0) | 4 (80.0) | 0 (0.0) | 18 (66.7) | 1 (3.7) | 31 (62.0) | 2 (4.0) |
| Myalgia* | 1 (12.5) | 0 (0.0) | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 10 (37.0) | 2 (7.4) | 14 (28.0) | 2 (4.0) |
| Fatigue | 6 (75.0) | 1 (12.5) | 3 (30.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 8 (29.6) | 0 (0.0) | 17 (34.0) | 1 (2.0) |
| Pyrexia | 2 (25.0) | 2 (25.0) | 2 (20.0) | 0 (0.0) | 2 (40.0) | 0 (0.0) | 9 (33.3) | 2 (7.4) | 15 (30.0) | 4 (8.0) |
| Cough | 1 (12.5) | 0 (0.0) | 3 (30.0) | 0 (0.0) | 2 (40.0) | 0 (0.0) | 5 (18.5) | 0 (0.0) | 11 (22.0) | 0 (0.0) |
| Anemia | 5 (62.5) | 5 (62.5) | 4 (40.0) | 2 (20.0) | 4 (80.0) | 2 (40.0) | 17 (63.0) | 11 (40.7) | 30 (60.0) | 20 (40.0) |
| Thrombocytopenia | 3 (37.5) | 1 (12.5) | 2 (20.0) | 0 (0.0) | 3 (60.0) | 2 (40.0) | 7 (25.9) | 4 (14.8) | 15 (30.0) | 7 (14.0) |
| Diarrhea | 3 (37.5) | 0 (0.0) | 3 (30.0) | 0 (0.0) | 4 (80.0) | 0 (0.0) | 12 (44.4) | 3 (11.1) | 22 (44.0) | 3 (6.0) |
| Constipation | 2 (25.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 1 (20.0) | 0 (0.0) | 7 (25.9) | 0 (0.0) | 11 (22.0) | 0 (0.0) |
| Nausea | 3 (37.5) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 8 (29.6) | 0 (0.0) | 12 (24.0) | 0 (0.0) |
| Vomiting | 3 (37.5) | 0 (0.0) | 2 (20.0) | 0 (0.0) | 1 (20.0) | 0 (0.0) | 6 (22.2) | 1 (3.7) | 12 (24.0) | 1 (2.0) |
| Blood creatine phosphokinase increased* | 1 (12.5) | 0 (0.0) | 2 (20.0) | 2 (20.0) | 3 (60.0) | 1 (20.0) | 12 (44.4) | 6 (22.2) | 18 (36.0) | 9 (18.0) |
| Dysgeusia* | 3 (37.5) | 0 (0.0) | 4 (40.0) | 0 (0.0) | 3 (60.0) | 0 (0.0) | 9 (33.3) | 0 (0.0) | 19 (38.0) | 0 (0.0) |
| Alopecia* | 3 (37.5) | 0 (0.0) | 7 (70.0) | 0 (0.0) | 3 (60.0) | 0 (0.0) | 12 (44.4) | 0 (0.0) | 25 (50.0) | 0 (0.0) |
Associated with sonidegib treatment.
The most frequently reported AEs (≥20% of patients, all grades) suspected to be related to the study treatment are shown in supplemental Table 4. The most commonly reported AEs (≥10% of patients, grade 3/4) suspected to be related to the study treatment were increased blood creatine phosphokinase (18%), anemia (14%), and thrombocytopenia (12%).
SAEs were reported in 26 patients (52%). The most frequent SAEs (≥5% of patients, all grades) were pyrexia (10%) and increased blood creatine phosphokinase (10%).
In phase 1b and phase 2 stage 1, 29 patients (58%) discontinued the study drug due to AEs (all grades, regardless of the study drug relationship); most frequently reported AEs (≥5% of patients) leading to discontinuation were increased blood creatine phosphokinase (8%), thrombocytopenia (6%), muscle spasms (6%), AML (6%), and alopecia (6%). Thirty-six patients (72%) required dose adjustment or interruption due to AEs (all grades); most frequently reported AEs (≥5% of patients) were blood creatine phosphokinase increased (18%); thrombocytopenia (14%), anemia (12%), muscle spasms (10%), myalgia (10%), alopecia (10%), platelet count decreased (8%); and diarrhea (6%).
Four deaths were reported during the study due to AML, MF progression, multiple organ dysfunction syndrome, and aspiration pneumonia (1 patient each); of these, 1 death was on treatment (multiple organ dysfunction syndrome).
PK
The observed PK parameter values for ruxolitinib and sonidegib were comparable to those from historical clinical studies where the drugs were dosed as a single agent. Further details on PK are provided in supplemental Tables 5 and 6. Geometric mean and arithmetic mean concentration-time profiles by treatment arms at week 1, day 1 and week 9, day 1 for ruxolitinib and sonidegib are shown in supplemental Figure 3.
Early termination
The study was terminated early on 10 April 2018 (the last patient’s last visit). The benefit-risk assessment of the combination treatment as compared with historical ruxolitinib monotherapy based on interim analysis data was not supportive to pursue further development in MF patients.
Discussion
In this phase 1b/2, open-label, multicenter, dose-finding study, the MTD/RP2D of the combination of sonidegib with ruxolitinib in JAKi-naive patients with MF was determined to be sonidegib 400 mg daily and ruxolitinib 20 mg twice daily. The MTD of the combination comprised lower doses of both drugs compared with individual MTDs for sonidegib (800 mg daily) and ruxolitinib (25 mg twice daily).17,18
No unexpected DLTs were observed with the combination. In the current study, 3 patients experienced grade 3 or 4 increased blood creatine phosphokinase (2 patients [4%] at sonidegib 400 mg daily and ruxolitinib 15 mg twice daily and 1 patient [2%] at the MTD/RP2D dose from the phase 1b expansion part) as a DLT, which is consistent with the known profile of sonidegib.17 In a dose-escalation study conducted by Rodon et al, reversible grade 3 or 4 serum creatine kinase elevation was considered to be a DLT at doses ≥800 mg daily and ≥250 mg twice daily. Thrombocytopenia was defined as a DLT for ruxolitinib alone.18
The combination caused a modest reduction in spleen volume. In the phase 1b expansion and phase 2 stage 1 part of the study, 44.4% and 29.6% of patients achieved ≥35% reduction in spleen volume as per MRI/CT by weeks 24 and 48, respectively. In the COMFORT trials, 41.9% of patients at week 24 (COMFORT-I) and 28% of patients at week 48 (COMFORT-II) achieved ≥35% reduction in spleen volume with ruxolitinib alone.1,4 Therefore, the anticipated synergistic effect with the combination of sonidegib and ruxolitinib was not observed.
Significant differences were not observed in other markers of efficacy. Few patients showed improvement in BM fibrosis. The reduction was modest in MFSAF TSS and minimal in JAK2 V617F allele burden and Gli1 messenger RNA expression. Activation of Gli proteins is observed in several cancers, including BCC, malignant gliomas, leukemias, and carcinomas of the breast, lung, pancreas, and prostate.19 The Gli1 expression results in this study are inconclusive due to limited sample size. However, in the BOLT study in patients with locally advanced BCC, substantial and durable inhibition of Gli1 expression was observed with 200 mg and 800 mg sonidegib (>90% median decreases from baseline at weeks 9 and 17).20 In addition, potent Gli1 suppression (median 63-fold reduction from baseline) was observed with 800 mg daily sonidegib in men with high-risk localized prostate cancer undergoing radical prostatectomy.21
This trial was based on the preclinical murine BM transplant model of PMF showing significant efficacy of ruxolitinib and sonidegib combination therapy in reduction of splenomegaly, JAK2 allele burden, and BM fibrosis compared with ruxolitinib monotherapy. Unfortunately, we were unable to translate any of these additional benefits in this trial at a prespecified end point. Several novel agents in combination with ruxolitinib therapy have been investigated in MF with strong scientific rationale derived from preclinical models.22 To the best of our knowledge, none of these agents have reached phase 3 stage. This study highlights important caveat and limitations in translating the findings of animal model research in humans. Perhaps current animal models do not capture the clonal complexity of MF well, and better models are required to move the research in this field further.
The PK profile of ruxolitinib and sonidegib in this study was in accordance with those observed in previous studies where the drugs were dosed as single agents, suggesting combining sonidegib and ruxolitinib did not appear to affect the PK of either agent.
The safety profile of the combination was consistent with those of the individual drugs. In our study, the most commonly reported grade 3 or 4 AEs (≥10% of all patients, regardless of the study drug relationship) were anemia (40%), increased blood creatine phosphokinase (18%), and thrombocytopenia (14%). Based on previous reports, increased serum creatine kinase was one of the most commonly reported AEs (>10% of patients, grade 1 or 2) experienced with sonidegib, while anemia and grade 3 or 4 thrombocytopenia were the most common hematologic AEs associated with ruxolitinib as observed in the COMFORT trials.1,4,13,17,18 Other AEs observed in this study, including muscle spasms, myalgia, dysgeusia, and alopecia, were associated with sonidegib treatment, which is consistent with the findings of the BOLT trial.20 Furthermore, alopecia, muscle spasms, and dysgeusia have been observed with the recently approved Hh pathway inhibitor glasdegib. However, the frequencies of these AEs were numerically lower than in a previous report.12 In line with prior publications, the majority of these AEs were usually managed through dose reduction or interruption.17,18
In summary, Sonidegib 400 mg daily + ruxolitinib 20 mg twice daily was declared as the RP2D for the combination treatment. Although the combination treatment of sonidegib and ruxolitinib was generally well tolerated, with a safety profile consistent with those of the individual drugs, the overall benefit of the combination was relatively modest in comparison with ruxolitinib monotherapy. Due to the lack of sufficient efficacy of this combination, further investigations beyond the current study will not be pursued in patients with MF.
Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The availability of this trial data is according to the criteria and process described on www.clinicalstudydatarequest.com.
Acknowledgment
The authors thank Nupur Chaubey, Novartis Healthcare Pvt Ltd (Hyderabad, India) for her medical writing assistance with this manuscript.
Authorship
Contribution: S.K. has contributed to the evaluation and discussion of patients’ clinical responses and AEs, to the discussion of the results of the overall study, including PK, and to the medical review of this manuscript; Y.L. has contributed to the pharmacokinetic analysis, summary, and interpretation of this study; T.D. has contributed to the statistical analysis and interpretation of the results for the study; A.M.V., C.H., and F.C. contributed to enrolling patients in the trial, interpretation of results, and review of this manuscript; M.W. has contributed to the medical review of this manuscript; S.B. has contributed to the scientific design of the study and content review of this manuscript; D.W. and H.H. contributed to patient enrollment in the trial and review of this manuscript; and V.G. contributed to patient enrollment, data analysis, writing, and final review of the manuscript.
Conflict-of-interest disclosure: V.G. declares accepting grants from Novartis and Incyte; personal fees from Novartis, Incyte, Celgene, and Sierra Oncology; and other grants from serving on the advisory boards of Novartis, Celgene, and Sierra Oncology. A.M.V. reports personal fees from Novartis, CTI, Celgene, and Incyte outside of the submitted work. F.C. reports financial activity from the advisory boards of Novartis, Celgene, and Farmitalia; and as a member of the speakers bureaus at Novartis and Celgene. S.K. reports receiving grant, personal fees, nonfinancial support, and AdBoard and travel reimbursement from Novartis during the conduct of the study, and grant, personal fees, and AdBoard and travel reimbursement from BMS, AOP pharma, and Janssen; and personal fees and AdBoard and travel reimbursement from Incyte/Ariad, CTI, Roche, Sanofi, Shire, Celgene, and Bayer. Y.L., T.D., M.W., and S.B. report personal fees from Novartis as employees. In addition, M.W. and S.B. report stock incentives from Novartis. C.H. reports payment for research and grant from Novartis and personal fees from Novartis, AOP pharma, Celgene, Janssen, Sierra Oncology, and Promedior, and travel costs from CTI. H.H. reports grants from Novartis Healthcare A/S and provides consultation for AORPHAN. D.W. declares no competing financial interests.
Correspondence: Vikas Gupta, Princess Margaret Cancer Centre, University of Toronto, 610 University Ave, Toronto, ON M5G 2M9, Canada; e-mail: vikas.gupta@uhn.ca.

