

Key Points
MK-specific double deficiency of Twf1 and Cof1 causes macrothrombocytopenia, defective proplatelet formation, and impaired platelet function.
Twf1 and Cof1 have essential synergistic functions in the regulation of actin/microtubule crosstalk in MKs.

Abstract
Rearrangements of the microtubule (MT) and actin cytoskeleton are pivotal for platelet biogenesis. Hence, defects in actin- or MT-regulatory proteins are associated with platelet disorders in humans and mice. Previous studies in mice revealed that loss of the actin-depolymerizing factor homology (ADF-H) protein Cofilin1 (Cof1) in megakaryocytes (MKs) results in a moderate macrothrombocytopenia but normal MK numbers, whereas deficiency in another ADF-H protein, Twinfilin1 (Twf1), does not affect platelet production or function. However, recent studies in yeast have indicated a critical synergism between Twf1 and Cof1 in the regulation of actin dynamics. We therefore investigated platelet biogenesis and function in mice lacking both Twf1 and Cof1 in the MK lineage. In contrast to single deficiency in either protein, Twf1/Cof1 double deficiency (DKO) resulted in a severe macrothrombocytopenia and dramatically increased MK numbers in bone marrow and spleen. DKO MKs exhibited defective proplatelet formation in vitro and in vivo as well as impaired spreading and altered assembly of podosome-like structures on collagen and fibrinogen in vitro. These defects were associated with aberrant F-actin accumulation and, remarkably, the formation of hyperstable MT, which appears to be caused by dysregulation of the actin- and MT-binding proteins mDia1 and adenomatous polyposis coli. Surprisingly, the mild functional defects described for Cof1-deficient platelets were only slightly aggravated in DKO platelets suggesting that both proteins are largely dispensable for platelet function in the peripheral blood. In summary, these findings reveal critical redundant functions of Cof1 and Twf1 in ensuring balanced actin/microtubule crosstalk during thrombopoiesis in mice and possibly humans.
Introduction
Platelets are small anucleate cell fragments derived from giant precursor cells, the megakaryocytes (MKs), residing in the bone marrow (BM). Mature polyploid MKs extend long protrusions (“proplatelets”) into the sinusoidal vessel lumen, which are shed by shear forces and further fragment into platelets in the blood stream.1 Proplatelet formation (PPF) requires extensive microtubule (MT) and actin rearrangements,2,3 and defects in proteins regulating cytoskeletal dynamics are associated with platelet disorders in humans and mice.4-7
The role of MT for proplatelet elongation and transport of granules into nascent proplatelets is well established.8 On the other hand, the critical importance of actin dynamics in platelet biogenesis is emphasized by the identification of damaging mutations in genes encoding actin-binding proteins such as filamin A, actinin 1, and tropomyosin 4 in patients with macrothrombocytopenia and the corresponding knock-out mouse models.6,7,9 Furthermore, studies in mice have associated loss of the actin-binding proteins tropomodulin 3, profilin 1, and Arp2/3 with thrombocytopenia.10-12 However, the molecular mechanisms that orchestrate actin dynamics and mediate the crosstalk between actin and MT during platelet biogenesis are still incompletely understood.
One of the best-characterized actin-binding motifs is the ADF-H domain. Besides actin-depolymerizing factor (ADF) and Cofilin1 (Cof1), also twinfilins (Twfs), Abp1/drebrin, and coactosin belong to the ADF-H family of actin-binding proteins.13,14 ADF/Cof1 contain a single ADF-H domain and preferentially bind to ADP-F actin. Binding of ADF/Cof1 induces a twist in the filament, thereby promoting actin severing at pointed ends.15,16 Twfs are composed of 2 ADF-H domains connected by a short linker region followed by a C-terminal tail.17 In humans, 3 different Twf isoforms have been identified: the ubiquitously expressed Twf1 and Twf2a and the muscle-specific Twf2b.18,19 Twfs preferentially bind to monomeric ADP-bound G-actin, thus promoting sequestration of actin monomers.20,21 Moreover, Twfs prevent barbed end growth by capping of actin filaments22,23 and regulate actin disassembly by enhancing barbed end depolymerization.24,25
We have previously identified the small actin-binding proteins ADF and Cof1 as critical determinants of platelet formation and sizing in mice.26 Combined loss of ADF/Cof1 in MKs virtually abrogated actin dynamics resulting in defective PPF and dramatic macrothrombocytopenia demonstrating that functional actin dynamics are pivotal for platelet production from MKs.26 In contrast, the loss of Twf2a was shown to cause macrothrombocytopenia in mice because of markedly increased platelet clearance in the spleen.27 Platelets derived from Twf2a-deficient animals were hyperresponsive toward agonist-induced activation and displayed defects in actin dynamics as well as altered Profilin 1 and Cof1 activity, thus highlighting the inhibitory function of Twf2a during actin polymerization. In contrast, Twf1 deficiency did not affect platelet number or reactivity, suggesting that the protein is dispensable for platelet production and function.
Recent studies in yeast indicated that Twf1 and Cof1 may synergistically regulate actin dynamics.28,29 Twf deficiency led to mild alterations in the cortical actin patches and a defective budding pattern, whereas the combination of Twf deficiency with a Cof functional mutant resulted in synthetic lethality.20 In addition, a study by Moseley et al indicated a functional redundancy between Cof and Twf in budding yeast.29 Both proteins were shown to induce actin severing, thereby increasing the amount of free actin barbed ends. In contrast to Cof1, however, Twf1-dependent actin severing was inhibited in the presence of capping proteins or increasing amounts of monomeric actin. Whether Twf1 and Twf2a share similar functions as demonstrated for Twf1 and Cof1 has not been addressed, although it was shown that they display distinct expression patterns and appeared to be differentially regulated.18
In this study, we used knockout mouse models to further dissect the interplay and distinct functions of Twf1/2a and Cof1 in platelet biogenesis and function. We show that Twf1/2a double-deficient mice recapitulate the phenotype described for Twf2 deficiency, demonstrating largely nonoverlapping roles of Twf1 and Twf2a in MKs and platelets. In contrast, our results reveal that Twf1 and Cof1 have critical synergistic functions in orchestrating actin/microtubule crosstalk and remodeling during platelet biogenesis.
Materials and methods
Animals
All animal studies were approved by the district government of Lower Franconia (Bezirksregierung Unterfranken, AZ 55.2.2-2532-2-1021-29). Platelet- and megakaryocyte-specific conditional Cof1-deficient and Twf1-deficient mice were generated as described previously.26,27,30 Double-deficient animals (DKO) were generated by intercrossing Twf1fl/fl mice with Cof1fl/fl,Pf4Cre mice. Eight- to 12-week-old DKO mice and matching Cre-negative littermates were used for experiments. Twf1fl/fl,Pf4Cre are referred to as Twf1−/−, whereas Cof1fl/fl,Pf4Cre mice are referred to as Cof1−/−. Twf1/Twf2a double-deficient mice were generated by intercrossing Twf1fl/fl,Pf4Cre with Twf2a−/− mice and are further referred to as Twf1−/−/Twf2a−/−.
Detailed protocols for platelet preparation, determination of platelet lifespan, count and size, tail bleeding time, 2-photon microscopy, spreading assays and clot retraction, as well as MK differentiation and culture, histology, staining procedures, and immunoblotting can be found in supplemental Materials and methods.
Data analysis
The presented results are mean ± standard deviation (SD) from at least 3 independent experiments per group, if not stated otherwise. Data distribution was analyzed using the Shapiro-Wilk test and differences between control and knockout mice were statistically analyzed using unpaired, 2-tailed Student t test, 1-way analysis of variance (ANOVA) or Fisher’s exact test. P < .05 was considered statistically significant: *P < .05; **P < .01; ***P < .001. Results with P > .05 were considered as nonsignificant.
Results
Severe macrothrombocytopenia in Twf1/Cof1-double deficient mice
In contrast to the proposed synergism between Twf1 and Cof1 in yeast, no such report exists on Twf1 and Twf2a. We generated Twf1/Twf2a double-deficient (Twf1−/−/Twf2a−/−) mice and found that additional loss of Twf1 did not affect platelet and MK parameters beyond those described for Twf2a single deficiency. This was true for platelet count and size (supplemental Figure 1A-B) as well as platelet reactivity in vitro (supplemental Figure 1C-D). Also, MK numbers in the BM were similar to those in Twf2a single-deficient mice (supplemental Figure 1E) and in line with our previous findings,27 an increased abundance of GPIX-positive particles in spleens of Twf1−/−/Twf2a−/− mice indicated enhanced platelet clearance. Together, these results demonstrated largely nonredundant roles of these ADF-H proteins in MKs and platelets.
To investigate a potential interplay of Twf1 and Cof1, MK- and platelet-specific Twf1/Cof1 DKO mice were generated and born in the expected mendelian ratios (Figure 1A). In line with previous reports, platelet count and size were unaltered in Twf1−/− mice,27 whereas loss of Cof1 resulted in a moderate reduction in platelet counts associated with a pronounced increase in platelet size (Figure 1B, C). Remarkably, the platelet count was even further reduced in DKO mice (WT: 868 ± 159 × 103/µL, Twf1−/−: 926 ± 218 × 103/µL, Cof1−/−: 581 ± 78 × 103/µL, DKO: 414 ± 118 × 103/µL; Figure 1B; #P < .05), whereas the size of the DKO platelets was similar to Cof1−/− platelets. This was also evident in transmission electron microscope images (Figure 1D). Numbers of marginal MT coils, as well as α- and dense granules were not altered in DKO platelets compared to the control (Figure 1E-F).

Severe macrothrombocytopenia in MK-specific Twf1/Cof1-deficient mice. (A) Absence of Twf1 and Cof1 was confirmed in platelets derived from WT, Twf1−/−, Cof1−/−, and DKO mice by immunoblotting. Platelet count (B) and size (C) were determined using an automated blood cell analyzer (ScilVet). Values of Twf1−/−, Cof1−/−, and DKO mice were normalized to the respective WT control. Values for all controls are shown. Mean ± SD (n = 9). One-way ANOVA with Sidak correction for multiple comparisons. #P < .05; **P < .01; ***P < .001. (D) Transmission electron microscopic images of WT and DKO platelets. Scale bars, 0.5 µm. Insets display microtubule coils. Scale bars, 0.2 µm. Analysis of microtubule (E), dense granule (F), and α-granule (G) numbers in WT and DKO platelets. Values are mean ± SD (n = 3).
Severe macrothrombocytopenia in MK-specific Twf1/Cof1-deficient mice. (A) Absence of Twf1 and Cof1 was confirmed in platelets derived from WT, Twf1−/−, Cof1−/−, and DKO mice by immunoblotting. Platelet count (B) and size (C) were determined using an automated blood cell analyzer (ScilVet). Values of Twf1−/−, Cof1−/−, and DKO mice were normalized to the respective WT control. Values for all controls are shown. Mean ± SD (n = 9). One-way ANOVA with Sidak correction for multiple comparisons. #P < .05; **P < .01; ***P < .001. (D) Transmission electron microscopic images of WT and DKO platelets. Scale bars, 0.5 µm. Insets display microtubule coils. Scale bars, 0.2 µm. Analysis of microtubule (E), dense granule (F), and α-granule (G) numbers in WT and DKO platelets. Values are mean ± SD (n = 3).
In situ analysis of the ultrastructure of DKO MKs by transmission electron microscopy revealed fewer and irregular membrane invaginations suggesting that demarcation membrane system (DMS) maturation was affected in the absence of Twf1 and Cof1 (Figure 2A). Quantification of DMS dilation as a measure of impaired DMS maturation31 revealed a significant increase in loosened membrane structures in the DKO MKs (Figure 2B; supplemental Figure 2A). These membrane alterations were not observed in Cof1−/− MKs (supplemental Figure 2A). Femur cryosections from DKO mice stained for MKs (GPIX) and sinusoidal vessels (CD105/endoglin) revealed a dramatic increase in MK numbers (Figure 2B-C). In contrast, MK numbers in the BM of Cof1−/− or Twf1−/− mice were not altered compared with the respective control (Figure 2B-C). Analysis of hematoxylin and eosin-stained femora and spleen sections confirmed these results and further revealed increased MK numbers in spleens of DKO mice (supplemental Figure 2B-C). In line with the increased number of BM MKs, we detected an elevated amount of megakaryocyte-erythroid progenitors in the BM of DKO mice and a concomitant decrease in common myeloid progenitors (supplemental Figure 4A-B). MK ploidy levels in vivo were largely unaltered as compared to WT animals, except for a small, but significant increase in the proportion of 16N MKs in DKO mice (supplemental Figure 2D). Taken together, these findings indicated impaired DMS development but otherwise largely functional maturation of DKO MKs in the BM.

Altered MK morphology and increased number of bone marrow MKs in DKO mice. (A) BM MKs from WT and DKO mice were analyzed by transmission electron microscopy. Scale bars, 1 µm. (B) DMS dilation as a measure of altered DMS maturation was quantified using ImageJ software (National Institutes of Health). Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. ***P < .001. (C) Confocal fluorescence microscopic images of femora cryosections of WT, Twf1−/−, Cof1−/−, and DKO mice (Leica TCS SP5). Scale bars, 50 µm. MKs, proplatelets, and platelets are shown by GPIX staining in green. CD105 staining (red) labels vessels. Nuclei were counterstained using DAPI (blue). (D) Quantification of BM MKs in whole femora cryosections. Values of Twf1−/−, Cof1−/−, and DKO mice were normalized to the respective WT control. Values are mean ± SD (n = 3). One-way ANOVA with Sidak correction for multiple comparisons. **P < .01.
Altered MK morphology and increased number of bone marrow MKs in DKO mice. (A) BM MKs from WT and DKO mice were analyzed by transmission electron microscopy. Scale bars, 1 µm. (B) DMS dilation as a measure of altered DMS maturation was quantified using ImageJ software (National Institutes of Health). Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. ***P < .001. (C) Confocal fluorescence microscopic images of femora cryosections of WT, Twf1−/−, Cof1−/−, and DKO mice (Leica TCS SP5). Scale bars, 50 µm. MKs, proplatelets, and platelets are shown by GPIX staining in green. CD105 staining (red) labels vessels. Nuclei were counterstained using DAPI (blue). (D) Quantification of BM MKs in whole femora cryosections. Values of Twf1−/−, Cof1−/−, and DKO mice were normalized to the respective WT control. Values are mean ± SD (n = 3). One-way ANOVA with Sidak correction for multiple comparisons. **P < .01.
To elucidate whether the macrothrombocytopenia in DKO mice was influenced by enhanced platelet clearance from the circulation, we analyzed the in vivo platelet lifespan and found it to be unaltered as compared to the control (supplemental Figure 3A). However, platelet recovery upon anti-GPIbα antibody-mediated platelet depletion was significantly delayed in DKO mice (supplemental Figure 3B-C), thus pointing to a platelet production defect as the major cause of the thrombocytopenia in these animals.
Impaired proplatelet formation in the absence of Twf1 and Cof1
To further investigate the effect of Twf1/Cof1 double deficiency on platelet biogenesis, we analyzed the ability of BM-derived DKO MKs to form proplatelets in vitro. Although the size of mature cultured DKO MKs was unaltered compared with WT controls (supplemental Figure 4C), PPF was mildly reduced in the absence of both Twf1 and Cof1 (Figure 3A). Microscopic analysis of the MKs illustrated the presence of thickened and shortened proplatelet shafts, suggesting that a dysfunctional cytoskeleton accounted for the reduced PPF (supplemental Figure 4D). In contrast, PPF in vitro was not affected by single deficiency in either Twf1 or Cof1 (supplemental Figure 5).26,27 We further analyzed F-actin and microtubules by confocal immunofluorescence microscopy and could identify similarly thickened proplatelet shafts as observed by brightfield microscopy (Figure 3C). In addition, the amount of proplatelet extensions per MK was markedly reduced in the DKO compared with the control, whereas the size of proplatelet tips was significantly increased (Figure 3D), thus emphasizing that loss of Cof1 and Twf1 affected both proplatelet elongation and branching. To gain a deeper understanding of the dynamics of proplatelet generation of DKO MKs, we performed time-lapse microscopy of cultured BM MKs derived from WT (supplemental Video 1) and DKO (supplemental Video 2) mice. As visible in supplemental Video 2, the area of proplatelet-forming DKO MKs was markedly reduced in line with fewer protrusions compared to WT MKs.

Impaired proplatelet formation of DKO MKs in vitro and in vivo. (A) PPF of BM MKs after lineage depletion and culturing in rHirudin- and TPO-conditioned medium. Proplatelet-forming MKs were counted after enrichment of MKs using a body surface area density gradient. Average of 5 analyzed visual fields per MK culture is shown. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05. (B) Intravital 2-photon microscopy of BM MKs in the skull. Blood vessels were visualized with body surface area-FITC and CD105 Alexa F488. MKs were stained with an anti-GPIX antibody derivative conjugated to Alexa F546. Insets and arrows point to proplatelet shafts reaching into sinusoidal vessels. Images are representative of at least n = 5. Scale bars, 50 µm; insets, 20 µm. BM MKs were centrifuged onto glass slides. (C) Proplatelets were visualized using an α-tubulin antibody and phalloidin and analyzed by confocal microscopy (40× objective, Leica TCS SP8) using a 40× objective. Scale bars, 20 µm (upper panels) and 50 µm (lower panels). (D) Quantification of platelets tips per MK and platelet tip size of in vitro–matured WT and DKO MKs. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. ***P < .001.
Impaired proplatelet formation of DKO MKs in vitro and in vivo. (A) PPF of BM MKs after lineage depletion and culturing in rHirudin- and TPO-conditioned medium. Proplatelet-forming MKs were counted after enrichment of MKs using a body surface area density gradient. Average of 5 analyzed visual fields per MK culture is shown. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05. (B) Intravital 2-photon microscopy of BM MKs in the skull. Blood vessels were visualized with body surface area-FITC and CD105 Alexa F488. MKs were stained with an anti-GPIX antibody derivative conjugated to Alexa F546. Insets and arrows point to proplatelet shafts reaching into sinusoidal vessels. Images are representative of at least n = 5. Scale bars, 50 µm; insets, 20 µm. BM MKs were centrifuged onto glass slides. (C) Proplatelets were visualized using an α-tubulin antibody and phalloidin and analyzed by confocal microscopy (40× objective, Leica TCS SP8) using a 40× objective. Scale bars, 20 µm (upper panels) and 50 µm (lower panels). (D) Quantification of platelets tips per MK and platelet tip size of in vitro–matured WT and DKO MKs. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. ***P < .001.
We observed significantly decreased TPO levels in the plasma of DKO mice compared with WT controls (supplemental Figure 6A), which may be explained by the increased MK mass in the DKO mice. To investigate whether altered TPO signaling may account for the impaired PPF of DKO MKs, we analyzed TPO/Mpl-induced JAK2/STAT3 phosphorylation by performing automated western blotting on TPO-stimulated MKs. Although a significant phosphorylation of both proteins was detectable upon TPO treatment, levels of phosphorylated JAK2 and STAT3 were similar between WT and DKO MKs (supplemental Figure 6D-E), thus ruling out a defect in TPO signaling in the absence of Twf1 and Cof1. Consistently, JAK2 phosphorylation was unaltered in TPO-stimulated DKO platelets (supplemental Figure 6B-C).
To visualize PPF in vivo, we imaged the BM in the skull of WT and DKO mice by 2-photon intravital microscopy. As shown in supplemental Video 3, proplatelet-forming MKs in WT mice produced protrusions that reached into the vessel sinusoids and were subsequently rapidly shed off by the blood flow (Figure 3B). In contrast, large, abnormally thick protrusions were observed in DKO mice (Figure 3B, white arrow). These aberrant proplatelets mostly stayed attached to the MK throughout the observation period of up to 20 minutes (Figure 3B; supplemental Videos 4 and 5). Of note, Cof1−/− mice did not exhibit aberrant proplatelet protrusions in vivo (supplemental Video 6). In summary, these results illustrated how Twf1 and Cof1 are cooperatively involved in the orchestration of thrombopoiesis in vitro and in vivo.
Defective actin and microtubule dynamics in Twf1/Cof1-double deficient MKs
To investigate how Twf1 or Cof1 deficiency affected MK cytoskeletal remodeling, we assessed adhesion and morphology of in vitro-cultured MKs on fibrinogen or Horm collagen. Strikingly, DKO MKs exhibited a significant decrease in spreading area on both substrates (Figure 4A,C; supplemental Figures 7A-B and 8), which was accompanied by a marked increase in the amount of F-actin (Figure 4B; supplemental Figure 7C). In line with this, cultured, nonadherent DKO MKs displayed an increased amount of F-actin compared with the WT (supplemental Figure 9A). Strikingly, irregular F-actin accumulations were also observed in the periphery of DKO MKs in stained BM cryosections in situ (supplemental Figure 9B).
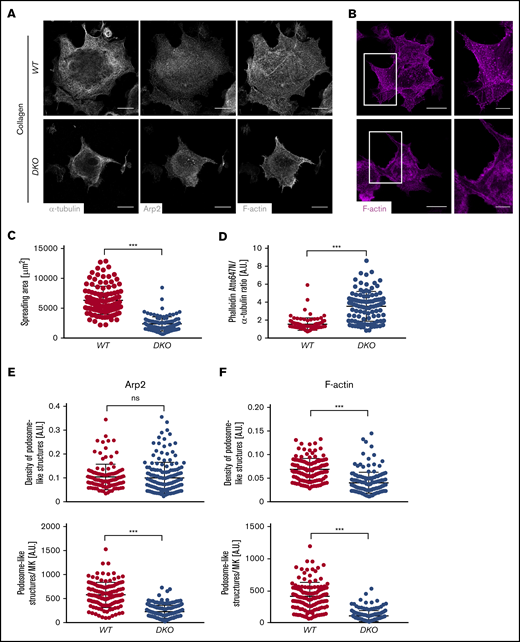
Defective spreading and actin remodeling of DKO MKs upon adhesion to Horm collagen. (A) Representative lowest plane images of spread BM-derived MKs analyzed by confocal microscopy (Leica TCS SP8) using a 40× objective. Scale bars, 25 µm. (B) Quantification of MK spreading area. At least 30 MKs were analyzed per animal. Values are mean ± SD (n = 3). (C-F) Analysis of F-actin distribution in WT and DKO MKs spread on Horm collagen. (B) Visualization of podosome-like structures by F-actin and Arp2 staining. Scale bars, 25 µm; insets, 10 µm. (C) Quantification of phalloidin fluorescence intensity (FI) normalized to α-tubulin (D), density of podosome-like structures (number per µm2) as well as total numbers in the Arp2- (E) and F-actin–channel (F) in control and DKO MKs spread on Horm collagen. Quantification was done using ImageJ Software. Unpaired, 2-tailed Student t test. ***P < .001. A.U., arbitrary unit; ns, nonsignificant.
Defective spreading and actin remodeling of DKO MKs upon adhesion to Horm collagen. (A) Representative lowest plane images of spread BM-derived MKs analyzed by confocal microscopy (Leica TCS SP8) using a 40× objective. Scale bars, 25 µm. (B) Quantification of MK spreading area. At least 30 MKs were analyzed per animal. Values are mean ± SD (n = 3). (C-F) Analysis of F-actin distribution in WT and DKO MKs spread on Horm collagen. (B) Visualization of podosome-like structures by F-actin and Arp2 staining. Scale bars, 25 µm; insets, 10 µm. (C) Quantification of phalloidin fluorescence intensity (FI) normalized to α-tubulin (D), density of podosome-like structures (number per µm2) as well as total numbers in the Arp2- (E) and F-actin–channel (F) in control and DKO MKs spread on Horm collagen. Quantification was done using ImageJ Software. Unpaired, 2-tailed Student t test. ***P < .001. A.U., arbitrary unit; ns, nonsignificant.
The correct assembly of podosome-like, F-actin–rich structures formed particularly during MK adhesion to collagen, was dramatically impaired upon analysis of the mean fluorescence of either the podosome marker Arp232 or the F-actin channel (Figure 4E-F). The density of podosome-like structures on the other hand was only affected when the distribution of F-actin was assessed. Of note, decreased spreading area (supplemental Figure 10A-B), increased F-actin content (supplemental Figure 10C-D) as well as reduced formation of podosome-like structures (supplemental Figure 10E-F) were to a lesser extent also observed in Cof1−/− MKs, whereas Twf1−/− MKs behaved largely comparable to the WT control. These results revealed that Twf1 and Cof1 have critical overlapping functions in the regulation of MK actin dynamics.
Because PPF elongation and abscission have been reported to be mainly driven by MT remodeling, we next assessed microtubule dynamics in DKO MKs. Posttranslational modifications by acetylation and detyrosination result in more long-lived, stable MT.33,34 Strikingly, we detected significantly increased levels of both acetylated and detyrosinated MT in DKO MKs (Figure 5A-B; supplemental Figure 7E), but not Cof1−/− or Twf1−/− MKs spread on collagen or fibrinogen (supplemental Figure 11). An increased proportion of detyrosinated MT was also observed in proplatelet-forming DKO MKs (supplemental Figure 12C-D) as well as in DKO platelets (supplemental Figure 15G), although acetylation was not significantly altered. Consistently, although treatment with the MT-disrupting toxin colchicine had no further effect on the defective spreading of DKO MKs, it diminished spreading of WT MKs to a similar extent as observed for the untreated DKO (Figure 5C; supplemental Figure 7F). Of note, colchicine treatment did not affect F-actin content or its altered distribution in DKO MKs. Despite the increased MT stability in DKO MKs, vesicle transport along MT appeared to be largely functional in the absence of Twf1/Cof1, as illustrated by the distribution of α granules (von Willebrand factor staining) within DKO and WT proplatelets (supplemental Figure 12A), in line with normal granule distribution in DKO platelets (Figure 1F-G; supplemental Figure 12B). Together, these results indicated that a combination of altered actin and MT dynamics was responsible for the impaired cytoskeletal rearrangements in DKO MKs.

Altered posttranslational modifications of microtubules in spread DKO MKs. (A) MKs were allowed to spread on Horm collagen and stained for acetylated or detyrosinated microtubules. (B) Fluorescence intensity of acetylated or detyrosinated microtubules was assessed using ImageJ Software. Values were normalized to α-tubulin. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. **P < .01; ***P < .001. (C) MKs were incubated with 500 µm colchicine for 30 minutes at 37°C, washed, resuspended in TPO-conditioned medium, and allowed to spread on Horm collagen. Spreading area was analyzed using ImageJ software. At least 30 MKs were analyzed per animal. Values are mean ± SD (n = 3). One-way ANOVA with Sidak correction for multiple comparisons. *P < .05; ***P < .001. colch, colchicine.
Altered posttranslational modifications of microtubules in spread DKO MKs. (A) MKs were allowed to spread on Horm collagen and stained for acetylated or detyrosinated microtubules. (B) Fluorescence intensity of acetylated or detyrosinated microtubules was assessed using ImageJ Software. Values were normalized to α-tubulin. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. **P < .01; ***P < .001. (C) MKs were incubated with 500 µm colchicine for 30 minutes at 37°C, washed, resuspended in TPO-conditioned medium, and allowed to spread on Horm collagen. Spreading area was analyzed using ImageJ software. At least 30 MKs were analyzed per animal. Values are mean ± SD (n = 3). One-way ANOVA with Sidak correction for multiple comparisons. *P < .05; ***P < .001. colch, colchicine.
Altered expression of proteins mediating actin-MT crosstalk in Twf1/Cof1 DKO platelets
Actin and MT dynamics are orchestrated by a plethora of proteins, some of which mediate crosstalk between the actin and the tubulin cytoskeleton. To better understand the mechanism underlying altered cytoskeletal dynamics in DKO MKs, we analyzed the expression and activity of selected actin and MT regulatory proteins by immunoblotting. In contrast to previous observations in Twf2a-deficient mice, we did not observe differences in Profilin1 phosphorylation in platelet lysates from DKO mice (supplemental Figure 13C). Moreover, protein levels of the Rho GTPase RhoA, a known key regulator of MK cytoskeletal dynamics,35,36 were not altered in DKO MKs compared with the control (Figure 6C-D). Notably, however, we detected reduced phosphorylation of the Cof1-regulating protein LIM Kinase 1 (LIMK1) in DKO platelets (supplemental Figure 13A-B). LIMK1 has been described to influence actin dynamics by phosphorylating Cof1 and to regulate MT stability,37,38 which is in line with the observed defects in DKO MKs. To further interpret the alterations in MT dynamics, we investigated protein and messenger RNA (mRNA) levels of several proteins reportedly coordinating both actin and MT rearrangements. Among these, we found diaphanous-related fomin-1 (mDia1) as well as adenomatous polyposis coli (APC) to be significantly downregulated on both mRNA (Figure 6A-B) and protein level in cultured DKO MKs (Figure 6C-D). Of note, we found similarly decreased Diaph1 and Apc mRNA levels in Cof1−/− MKs (supplemental Figure 14). Notably, the reduction in APC protein was only detectable in DKO MKs (Figure 6D), suggesting that additional degradation of the protein might occur in the DKO. Because both proteins have been implicated in platelet production and the regulation of MT stability in fibroblasts,5,34,39,40 differences in their protein abundance might explain the altered MT modifications in DKO MKs. In contrast to the reduction observed in DKO MKs, protein levels of mDia and APC were unaltered in DKO platelets (Figure 6D). Taken together, these findings revealed that altered expression of both actin and tubulin regulatory proteins resulted in increased MT stability and F-actin content in DKO MKs, translating into impaired MK spreading in vitro and reduced PPF in vivo.
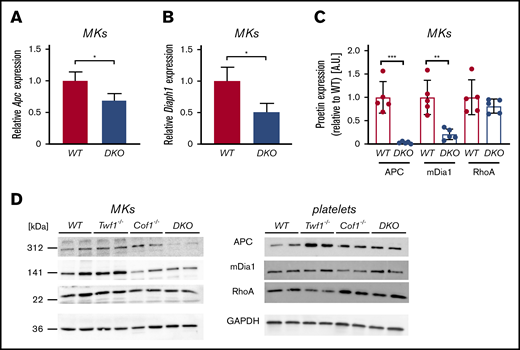
Diminished APC and mDia1 expression in DKO MKs, but not platelets. mRNA content of Apc (A) and Diaph1 (B) in in vitro-matured MKs was quantified by quantitative polymerase chain reaction. Levels were normalized to Sdha and Actb. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05. (C-D) MKs or washed platelets from WT, Twf1−/−, Cof1−/−, or DKO mice were immunoblotted for the cytoskeletal regulatory proteins APC, mDia1, and RhoA (D). Images were acquired using an Amersham Image 680 (GE Healthcare). Blots are representative of 3 independent experiments. (C) Densitometric analysis was performed using ImageJ Software. Values were normalized to WT levels. Values are mean ± SD. Unpaired, 2-tailed Student t test. **P < .01.
Diminished APC and mDia1 expression in DKO MKs, but not platelets. mRNA content of Apc (A) and Diaph1 (B) in in vitro-matured MKs was quantified by quantitative polymerase chain reaction. Levels were normalized to Sdha and Actb. Values are mean ± SD (n = 3). Unpaired, 2-tailed Student t test. *P < .05. (C-D) MKs or washed platelets from WT, Twf1−/−, Cof1−/−, or DKO mice were immunoblotted for the cytoskeletal regulatory proteins APC, mDia1, and RhoA (D). Images were acquired using an Amersham Image 680 (GE Healthcare). Blots are representative of 3 independent experiments. (C) Densitometric analysis was performed using ImageJ Software. Values were normalized to WT levels. Values are mean ± SD. Unpaired, 2-tailed Student t test. **P < .01.
DKO platelets display mild functional defects resembling Cof1-deficient platelets
Finally, we investigated the effect of combined Twf1/Cof1 deficiency on platelet function (supplemental Figure 15). Notably, the defects observed in DKO platelets were overall comparable to those described for Cof1−/− platelets26 (supplemental Figure 14), whereas Twf1 was dispensable for platelet function under all tested experimental conditions.27 Similarly to MKs, F-actin content was increased in unstimulated DKO platelets, whereas activation-induced F-actin assembly was impaired (supplemental Figure 15C-D). Spreading of DKO or Cof1−/− platelets on fibrinogen in vitro was delayed compared with the control and associated with the appearance of prominent MT structures at 15 minutes (supplemental Figures 14E and 15F). Colchicine treatment rescued the delayed spreading of DKO platelets, which is in concordance with the findings in MKs (Figure 5C; supplemental Figure 15F). In line with Cof1−/− platelets (supplemental Figure 14C-D), in vitro activation and degranulation were mildly impaired in DKO platelets in response to standard platelet agonists (supplemental Figure 15A-B). Ultimately, the mild activation defects translated into prolonged bleeding times in both DKO and Cof1−/− mice compared with the respective WT control (supplemental Figures 14F and 15E). Together, these results showed that the platelet phenotype in DKO mice was dominated by the loss of Cof1 and that, contrary to their importance in MKs, Twf1 and Cof1 are only to a limited extent required for platelet function.
Discussion
Our results reveal that Twf1 does not share functions with its homologue Twf2a, but is critically redundant to Cof1 in both actin and microtubule remodeling in MKs during platelet biogenesis. In contrast, platelet function is only moderately affected in the absence of Twf1 and Cof1.
It is proposed that the actin cytoskeleton is responsible for proplatelet branching, thereby increasing the number of available proplatelet tips.8,11 In line with this notion, Twf1/Cof1-deficient MKs formed proplatelets with few short branches and MKs derived from DKO mice exhibited increased F-actin content in vitro and in situ. Furthermore, the formation of podosome-like structures, which have been suggested to promote PPF through the basement membrane in vivo,32,41 was impaired in DKO MKs adherent to both collagen and fibrinogen matrices, albeit unaltered Arp2 distribution. Although the existence and relevance of MK podosomes in vivo is not firmly established, these observations may at least in part explain the platelet biogenesis defect of DKO and, to a lesser extent, Cof1-deficient MKs in vivo.
The actin-modulating functions of both Cof1 and Twf1 have been described in other mammalian cell types, as well as in yeast and drosophila. Unexpectedly, we furthermore observed strongly defective MT organization in DKO MKs, but not Twf1 or Cof1 single-deficient MKs. This was most evident in vivo as a thickening of proplatelet protrusions, which were extremely stable and therefore not shed off by the blood stream compared with the fast release of proplatelets observed by 2-photon microscopy in WT mice, a finding additionally supported by the aberrant morphology of DKO proplatelets observed by live imaging. Well-established indicators of increased MT stability are posttranslational acetylation and detyrosination.42 Although acetylation is a marker of MT stabilization in domains of slow turnover,43 detyrosination rather reflects longevity of MT subsets.44 We found increased amounts of detyrosinated MT in adherent and proplatelet-forming DKO MKs, as well as in DKO platelets, indicating that lack of Twf1 and Cof1 leads to the formation of hyperstable MT. Strikingly, the defective spreading of both DKO MKs and platelets was rescued to WT levels in the presence of the MT disrupting toxin colchicine, strongly suggesting that increased MT stability contributed not only to the spreading defect in DKO MKs in vitro, but also to the formation of hyperstable proplatelets in vivo. Because in vitro-matured MKs exhibited a similarly aberrant proplatelet morphology, these results strongly suggest that both impaired actin dynamics as well as increased MT stability account for the thrombocytopenia in Twf1/Cof1 double-deficient mice.
Of the numerous MT-regulating proteins, we detected markedly decreased expression of APC and mDia1 in DKO MKs on both protein and mRNA level. This suggests that transcription or mRNA stability, as well as protein turnover is affected in DKO MKs. Strikingly, APC and mDia1 have also been identified as regulators of platelet production in humans and mice. Although APC has been described as a negative regulator of PPF in a murine knockout model,39 global knockout of mDia1 in mice did not influence platelet numbers or reactivity,45 whereas its knockdown increased proplatelet formation in human MKs46 indicating possible species-specific differences in protein function. The decrease in protein content albeit impaired PPF in MKs lacking both Twf1/Cof1 therefore suggests that not the mere lack of the proteins, but rather a disrupted equilibrium of several proteins regulating actin/tubulin crosstalk, leads to the observed phenotype. In line with this, Twf1/Cof1 double deficiency resulted in a significantly reduced phosphorylation of the Cof1-regulator LIMK1, which has likewise been shown to regulate MT stability.38
The rather normal levels of mDia1 and APC may at least partially explain why only mild platelet function defects were observed in DKO platelets, which mostly reflected those found in Cof1-deficient platelets.26 The unaltered protein levels in circulating platelets further suggest an uneven distribution of proteins in DKO MKs, most likely because of the aberrant F-actin organization, and that only the MK cytoplasm containing reasonable amounts of mDia1 and APC is preferentially converted into platelets in vivo. Overall, our findings indicate that although Twf1 and Cof1 are key regulators of platelet biogenesis, they appear to be less critical for the hemostatic function of platelets in the peripheral blood. It is noteworthy that in both MKs and platelets, Cof1 single deficiency alone already resulted in mild defects, whereas Twf1 single-deficient MKs and platelets were fully functional in all tested assays.27 This apparent functional dominance of Cof1 may at least in part be explained by findings of proteomic studies demonstrating that Cof1 with >200 000 copies per platelet is among the most abundant proteins in human and mouse platelets, whereas Twf1 is expressed at much lower levels.47,48
To date, no direct association of variants in the twinfilin-1 (TWF1) or cofilin-1 (CFL1) genes with altered platelet counts has been described in humans. On the other hand, altered Cof1 phosphorylation has been observed in several thrombocytopenic mouse lines with deficiencies in cytoskeletal-regulatory genes, including Cdc42 and Pak2, classic upstream regulators of Cof1 activity,49,50 as well as Twf2a and Pdk1.27,51 Furthermore, aberrant regulation of Cof1 activity was functionally associated with thrombocytopenia in a mouse model for von Willebrand disease type 2B,52 as well as with defective PPF in MKs from a patient with platelet-type von Willebrand disease.53
Together, these findings emphasize the importance of Cof1 as a master regulator of cytoskeletal dynamics in MKs. Moreover, our study reveals critical overlapping functions of Twf1 and Cof1 in MKs and sheds new light on the importance of balanced actin-MT crosstalk during the process of platelet biogenesis.
Send data sharing requests via e-mail to the corresponding author, Bernhard Nieswandt (bernhard.nieswandt@virchow.uni-wuerzburg.de).
Acknowledgments
The authors thank Stefanie Hartmann and Sylvia Hengst for excellent technical assistance and the microscopy platform of the Bioimaging Center (Rudolf Virchow Centre) for providing technical infrastructure and support.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (NI 556/11-2 [B.N.] and project number 374031971–TRR 240/project A01) and the European Union (EFRE–Europäischer Fonds für regionale Entwicklung, Bavaria). I.C.B. was supported by a grant from the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg. M.B. is supported by an Emmy Noether grant of the Deutsche Forschungsgemeinschaft (BE5084/3-1).
Authorship
Contribution: I.C.B. and I.S. designed research, performed experiments, analyzed data, and wrote the manuscript; L.M.W., S.B., T.H., K.A., G.M., C.G., M.S., and Z.N. performed experiments and analyzed data; W.W. and P.L. provided mice and vital reagents; M.B. and H.S. commented on the manuscript; and I.P. and B.N. designed and supervised research, analyzed data, and wrote the manuscript.
Correspondence: Bernhard Nieswandt, Institute of Experimental Biomedicine, University Hospital and Rudolf Virchow Center, University of Würzburg; Josef-Schneider-Str 2, 97080 Würzburg, Germany; e-mail: bernhard.nieswandt@virchow.uni-wuerzburg.de.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

