

Key Points
CIT is upregulated in multiple myeloma.
CIT inhibition leads to cytokinesis failure and reduction in tumor burden in vitro and in vivo.

Abstract
Citron Rho-interacting serine/threonine kinase (CIT) is a serine/threonine kinase that acts as a key component of the midbody and is essential for cytokinesis. CIT has been reported to be highly expressed in some tumor tissues and to play a role in cancer proliferation; however, the significance of CIT has not been investigated in multiple myeloma (MM). Here, we identified, by protein microarray and immunohistochemistry, that CIT is 1 of the upregulated proteins in the plasma cells of MM patients compared with healthy controls. Analysis of a gene expression profile data set showed that MM patients with high CIT gene expression had significantly worse overall survival compared with MM patients with low CIT gene expression. CIT silencing in MM cell lines induced cytokinesis failure and resulted in decreased MM cell proliferation in vitro and in vivo. TP53 expression was found to be an independent predictor of CIT dependency, with low-TP53 cell lines exhibiting a strong dependency on CIT. This study provides the rationale for CIT being a potential therapeutic target in MM in future trials.
Introduction
Cytokinesis is the process, during cell division, that controls the proper partitioning of cytoplasmic and nuclear material between the 2 daughter cells. This process includes a series of sequential events. The midbody, a transient complex formed between 2 daughter cells at the end of cytokinesis, regulates the final abscission of the 2 cells.1
Citron Rho-interacting serine/threonine kinase (CIT) is highly enriched in the midbody of dividing cells and was found to be a key regulator of the molecular network required for cytokinesis.2,3 Therefore, inhibition of CIT may disrupt cytokinesis and cell growth. CIT is also found to be a cell cycle–dependent nuclear protein, which is required for the G2/M transition.4 It has been reported that CIT is overexpressed in several carcinomas compared with their normal-tissue counterparts.5,6 However, the significance of CIT expression in hematological malignancies, particularly multiple myeloma (MM), has not been well studied.
Despite recent therapeutic advances in MM, it is still an incurable disease and requires novel therapeutic approaches. Here, we identified CIT as 1 of the upregulated proteins in the plasma cells of MM patients compared with healthy controls by protein microarray. We analyzed the significance of CIT expression in MM using publicly available gene expression data sets and by studying bone marrow (BM) biopsy specimens. CIT-knockdown studies in MM cell lines were also performed to dissect the role of CIT in MM growth in vitro and in vivo. Our results suggest that CIT might be an important player in the pathogenesis of MM and a potential target in future myeloma trials.
Materials and methods
Protein microarray
An Antibody Microarray (BD Clontech, Palo Alto, CA) was conducted in 12 MM patients’ BM plasma cells. As previously described,7 the Antibody Microarray detects a wide variety of proteins (cytosolic and membrane bound) representing a broad range of biologic functions, including cell cycle, apoptosis, signal transduction, and gene transcription. The microarray contains 512 highly specific and sensitive monoclonal antibodies against human polypeptides.
Cells
The human MM cell lines MM.1s and OPM2 were purchased from the American Type Culture Collection (Manassas, VA). Cells were cultured in RPMI 1640 medium containing 2 mM/mL l-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin with 10% fetal bovine serum.
Generation of stable shRNA-expressing cell lines
Lentivirus supernatants of interest were obtained from The RNAi Consortium. As a control, the short hairpin RNA (shRNA) scramble vector was used, which contains an shRNA expression cassette with a target sequence not corresponding to any known mouse or human genes. The sequence of each shRNA is shown in supplemental Table 1. MM.1s and OPM2 cells were transduced with virus supernatants and 8 μg/mL Polybrene (hexadimethrine bromide; St. Louis, MO) and selected with puromycin 24 hours after the transduction. The efficacy of CIT knockdown was assessed by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR), as previously described.8
Gene expression analysis
CIT (Probe ID 212801_at) gene expression was analyzed using public data sets (GSE64779 and GSE265810 ). The R2 visualization platform (http://r2.amc.nl) was used to evaluate the prognostic role of the dysregulated expression of CIT in MM, as assessed by the Kaplan-Meier method in a large data set of newly diagnosed MM patients (N = 542; GSE2658). Median modus was used for cutoff determination, and the R2 platform was used for calculations.
Dependency Map analysis
Dependency Map11 (v18Q4) genome-wide CRISPR screen Ceres scores were interrogated for CIT dependency in available MM cell lines, and its association with gene expression and/or mutation and copy number status was explored using Cancer Cell Line Encyclopedia (CCLE) data.12 CCLE RPKM values for gene expression were transformed as follows: log2 (RPKM +1).
Immunohistochemistry
BM biopsy specimens from 11 MM patients and 6 healthy donors were fixed in formalin, embedded in paraffin blocks, and sectioned. Sections were stained with anti-CIT antibody (Sigma-Aldrich, St. Louis, MO).
Cell-proliferation assay and cell cycle analysis
Immunofluorescence
Immunofluorescence was performed as reported previously14 using DAPI nuclear stain (Vector Laboratories, Burlingame, CA) and α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA) and analyzed using a fluorescent microscope (Nikon Eclipse 80i). For the quantification of cells with cytokinesis failure, the number of multinucleated cells in random fields was counted at 100× magnification by 2 independent investigators and expressed as a percentage. At least 100 cells were evaluated per sample, with 3 independent replicates.
Immunoblotting
MM cells (MM.1s and OPM2) were cultured, harvested, and lysed. Proteins were detected by immunoblotting. Proteins were blotted using 8% to 12% acrylamide gels and transferred to a nitrocellulose membrane; membranes were blocked with 5% nonfat dry milk in TBS-Tween buffer and incubated with primary antibodies for anti-CIT antibody (Abcam), and α-tubulin as loading control (Cell Signaling Technologies), overnight at 4°C. The membranes were then washed, incubated with an appropriate horseradish peroxidase–conjugated secondary antibody, washed, and developed using a luminol-based assay. Luminescence was measured using radiographic films.
In vivo studies
Animal studies were approved by the Dana-Farber Cancer Institute Institutional Animal Care and Use Committee. Female 6- to 8-week-old SCID-Beige mice (Taconic, Hudson, NY) were injected subcutaneously into the left flank with 5 × 106 scramble control OPM2 cells or CIT-knockdown (clone F5) OPM2 cells (n = 8 and n = 7, respectively). Tumor growth was measured and calculated as (length [mm] × width2 [mm2])/2.
Statistical analysis
The statistical significance of differences between groups was determined using the Student t test. The minimal level of significance was P < .05. Survival data were generated using Kaplan-Meier survival analysis and the log-rank test.
Results
CIT is highly expressed in MM
Proteomic analysis using previously reported antibody-based protein arrays7 was performed on tumor cells from 12 patients with newly diagnosed MM; it identified CIT as 1 of the upregulated proteins in plasma cells from MM patients compared with healthy controls (Figure 1A). Using CIT (Probe ID 212801_at) gene expression data from public datasets (GSE6477),9 we also confirmed that the level of CIT expression was significantly higher in newly diagnosed MM patients' plasma cells compared to normal healthy individuals' plasma cells. Similarly, relapsed MM patients' plasma cells had higher CIT expression compared with newly diagnosed patients' samples (Figure 1B). Furthermore, BM biopsy specimens from 11 MM patients and 6 healthy donors were stained with anti-CIT antibody, which revealed higher CIT expression in MM samples compared with healthy donors (Figure 1C-D). Given the fact that CIT is highly expressed in MM cells compared with normal plasma cells, we next analyzed the gene-expression profiling data set to evaluate whether there is a correlation between CIT expression and prognosis in MM patients; better survival was seen in patients with low CIT expression (P = .01) (Figure 1E). These results imply that CIT is highly expressed in MM; therefore, targeting this kinase might provide a survival benefit in MM.
![Figure 1. CIT is highly expressed in MM patients. (A) Significant increase in CIT in primary BM-derived CD138+ MM plasma cells compared with healthy subjects by proteomic analysis using the Antibody Microarray (BD Clontech). (B) CIT gene expression levels in healthy control, newly diagnosed, and relapsed MM patients are shown. (C) Expression of CIT in BM biopsy specimens from MM patients (n = 11) and healthy donors (normal bone marrow [NMB]; n = 6) was detected by immunohistochemistry. Representative slides are shown for each group (original magnification ×20 [left] and ×60 [right]). (D) CIT+ cells in immunohistochemistry-stained slides were quantified using ImageJ software (P = .02). (E) Using CIT median gene expression value as a bifurcating point in GSE2658, Kaplan-Meier survival proportions analysis of patient outcome data demonstrates high levels of CIT gene expression associated with inferior OS (P = .01).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/7/10.1182_bloodadvances.2018028456/2/m_advances028456f1.png?Expires=1770926912&Signature=l~TO6e7WWWBPOPXK1Vvs93c-44WIosfkYi43ZdJhoXXBIJIsDxbDP0TmJEMOr2d1oqsappMFAY8l~NapDpKOw9eXHqbBC~nqjqgOEn28BnQ~BH2~B0QMJmeVNsWnTpc5wKBm3o3aSJ22NWt6~KbI6BPlVGhR2HKlz~Wh6Z1Qif3wK-gYAkiMUqWDTtTlhP2QV5v~fMBw2kFJ6LUg2Abg-qmTkzmhxLqrzc8pCOUGuFsudFoghs36~GK3Eu4XMECGuc65WHNPG~g2RQvDi6BW~janxKWOkhHhoUuudp6oHM1KtAnUleJC3XBQ37M5sj9QnKmaqfNFxzOkMpNFTP7~lg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CIT is highly expressed in MM patients. (A) Significant increase in CIT in primary BM-derived CD138+ MM plasma cells compared with healthy subjects by proteomic analysis using the Antibody Microarray (BD Clontech). (B) CIT gene expression levels in healthy control, newly diagnosed, and relapsed MM patients are shown. (C) Expression of CIT in BM biopsy specimens from MM patients (n = 11) and healthy donors (normal bone marrow [NMB]; n = 6) was detected by immunohistochemistry. Representative slides are shown for each group (original magnification ×20 [left] and ×60 [right]). (D) CIT+ cells in immunohistochemistry-stained slides were quantified using ImageJ software (P = .02). (E) Using CIT median gene expression value as a bifurcating point in GSE2658, Kaplan-Meier survival proportions analysis of patient outcome data demonstrates high levels of CIT gene expression associated with inferior OS (P = .01).
CIT is highly expressed in MM patients. (A) Significant increase in CIT in primary BM-derived CD138+ MM plasma cells compared with healthy subjects by proteomic analysis using the Antibody Microarray (BD Clontech). (B) CIT gene expression levels in healthy control, newly diagnosed, and relapsed MM patients are shown. (C) Expression of CIT in BM biopsy specimens from MM patients (n = 11) and healthy donors (normal bone marrow [NMB]; n = 6) was detected by immunohistochemistry. Representative slides are shown for each group (original magnification ×20 [left] and ×60 [right]). (D) CIT+ cells in immunohistochemistry-stained slides were quantified using ImageJ software (P = .02). (E) Using CIT median gene expression value as a bifurcating point in GSE2658, Kaplan-Meier survival proportions analysis of patient outcome data demonstrates high levels of CIT gene expression associated with inferior OS (P = .01).
The relative gene expression level of CIT messenger RNA in different MM cell lines, using the CCLE,12 was examined; the results showed similar expression levels (supplemental Figure 1A). We then examined CIT protein levels in several MM cell lines by immunoblotting and confirmed the expression at the protein level (supplemental Figure 1B).
CIT silencing reduces MM tumor growth in vitro and induces cytokinesis failure
To dissect the role of CIT in cytokinesis in MM, silencing of CIT in MM cell lines (OPM2 and MM.1s) was conducted using a lentiviral system (shRNA sequences; Scr, F5, and F10 are listed in supplemental Table 1), and knockdown of CIT was confirmed by qRT-PCR (primer sets and genes are listed in supplemental Table 2) (Figure 2A). CIT-knockdown cells exhibited a significant reduction in cell proliferation at 48 hours compared with scramble shRNA–infected cells of the OPM2 and MM.1s cell lines, as shown by [3H]-thymidine uptake assay15 (Figure 2B). To confirm whether CIT knockdown also disrupts cytokinesis in MM, immunofluorescence, using DAPI nuclear stain and α-tubulin, was performed to quantify cytokinesis failure. Significantly higher amounts of multinucleated cells, indicating cytokinesis failure, were observed in CIT-knockdown cells of the OPM2 and MM.1s cell lines compared with the scramble control (Figure 2C-D). Moreover, we examined the effect of CIT silencing on the cell cycle and compared the cell cycle phases with flow cytometry; we found a prolonged G2 cell cycle arrest in CIT-knockdown OPM2 cells compared with scramble control (Figure 2E), whereas MM.1s scramble control and CIT-knockdown cells had similar G2 cell cycle profiles (Figure 2E).
![Figure 2. CIT silencing decreases MM tumor growth in vitro and induces cytokinesis failure. (A) Depletion of CIT induced by shRNA (F5 and F10) in OPM2 and MM.1s cells was validated using qRT-PCR. (B) OPM2 and MM.1s CIT-knockdown cells were cultured for 48 hours. Cell proliferation was measured using [3H]-thymidine uptake assay. CIT silencing led to significant inhibition of MM cell proliferation (P < .001 and P < .02, respectively). (C) Immunofluorescence of OPM2 and MM.1s cells transfected with scramble or CIT shRNA (F5). Green arrows in enlarged images (magnification ×200) of original magnifications (×100) show nuclei; DAPI (blue) and α-tubulin (red). (D) Multinucleated OPM2 cells were quantified by immunofluorescence. Random fields were analyzed. A significant increase in multinucleated cells in short hairpin (sh)–CIT (F5)-knockdown cells was observed compared with scramble control. (E) The cell cycle of CIT-knockdown OPM2 and MM.1s cells was assessed by propidium iodide (PI) staining and flow cytometric analysis. Although silencing of CIT increased the G2-phase population in OPM2 cells, it did not have any effect on the G2-phase population in MM.1s cells. K.D., knockdown; scr/Scr, scramble.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/7/10.1182_bloodadvances.2018028456/2/m_advances028456f2.png?Expires=1770926912&Signature=jndPh7tl4zqW9HZP1uTTxpVPaJELiFF5QqWvBCj6akdJDSViS8nsJTHhMbtI0Koq-f1s2uPRNNVhRnjQzZFhfxFSa84V3FuW0o14toIub~MXABYjJ9RpY-8dgTE0GfqUc2mVsEWLHpsca3b3Yupg~4rC74gOK9nKVoXDo6mhIGp~XHYgIUpqDhDBxGlJqICTiluHpA421FDFKvaG-WSmSDzBomZVHSoOt-7w6tK3ImzTYJSlJ3nhSfAUSrUT0tlC3l17GHhPb8FiHkK6B~GzadvWgcVFo0Iptd2EnA5LguRFFwk3AHVurdCpFlth-9GaAuXNmpD6bNd0JPAarApRFg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CIT silencing decreases MM tumor growth in vitro and induces cytokinesis failure. (A) Depletion of CIT induced by shRNA (F5 and F10) in OPM2 and MM.1s cells was validated using qRT-PCR. (B) OPM2 and MM.1s CIT-knockdown cells were cultured for 48 hours. Cell proliferation was measured using [3H]-thymidine uptake assay. CIT silencing led to significant inhibition of MM cell proliferation (P < .001 and P < .02, respectively). (C) Immunofluorescence of OPM2 and MM.1s cells transfected with scramble or CIT shRNA (F5). Green arrows in enlarged images (magnification ×200) of original magnifications (×100) show nuclei; DAPI (blue) and α-tubulin (red). (D) Multinucleated OPM2 cells were quantified by immunofluorescence. Random fields were analyzed. A significant increase in multinucleated cells in short hairpin (sh)–CIT (F5)-knockdown cells was observed compared with scramble control. (E) The cell cycle of CIT-knockdown OPM2 and MM.1s cells was assessed by propidium iodide (PI) staining and flow cytometric analysis. Although silencing of CIT increased the G2-phase population in OPM2 cells, it did not have any effect on the G2-phase population in MM.1s cells. K.D., knockdown; scr/Scr, scramble.
CIT silencing decreases MM tumor growth in vitro and induces cytokinesis failure. (A) Depletion of CIT induced by shRNA (F5 and F10) in OPM2 and MM.1s cells was validated using qRT-PCR. (B) OPM2 and MM.1s CIT-knockdown cells were cultured for 48 hours. Cell proliferation was measured using [3H]-thymidine uptake assay. CIT silencing led to significant inhibition of MM cell proliferation (P < .001 and P < .02, respectively). (C) Immunofluorescence of OPM2 and MM.1s cells transfected with scramble or CIT shRNA (F5). Green arrows in enlarged images (magnification ×200) of original magnifications (×100) show nuclei; DAPI (blue) and α-tubulin (red). (D) Multinucleated OPM2 cells were quantified by immunofluorescence. Random fields were analyzed. A significant increase in multinucleated cells in short hairpin (sh)–CIT (F5)-knockdown cells was observed compared with scramble control. (E) The cell cycle of CIT-knockdown OPM2 and MM.1s cells was assessed by propidium iodide (PI) staining and flow cytometric analysis. Although silencing of CIT increased the G2-phase population in OPM2 cells, it did not have any effect on the G2-phase population in MM.1s cells. K.D., knockdown; scr/Scr, scramble.
Silencing of CIT inhibits MM tumor growth in vivo
To further validate the effect of CIT knockdown in MM cells, we next performed tumor xenograft experiments. MM cells (5 × 106 cells per SCID-Beige mouse) were injected subcutaneously into the left flank with either scramble control (n = 8) or CIT-knockdown cells (n = 7). Mice were followed for tumor growth and survival. Consistent with the in vitro data showing reduced proliferation in CIT-knockdown cells, significantly reduced tumor growth and prolonged survival were observed in mice injected with CIT-knockdown cells compared with mice injected with scramble control cells (Figure 3A-B). A decrease in CIT expression in the tumor cells from mice injected with CIT-knockdown cells was also confirmed ex vivo by qRT-PCR, showing that the injected cells maintained CIT knockdown (Figure 3C).
![Figure 3. CIT knockdown of MM cells reduced tumor growth in vivo. SCID/Beige mice were injected subcutaneously with OPM2 CIT-knockdown cells (n = 8) or scramble cells (n = 7) (5 × 106 per mouse) to establish an MM xenograft model. Tumor growth was measured, and volume was calculated as (length [mm] × width2 [mm2])/2. (A) CIT-knockdown cells showed reduced tumor growth in vivo (P < .001). (B) Mice bearing CIT-knockdown cells exhibited significantly prolonged survival (P < .001). (C) CIT expression of tumors collected from mice injected with scramble or CIT-knockdown cells. Decrease in CIT expression was confirmed ex vivo on harvested tumors using qRT-PCR. K.D, knockdown; Scr, scramble control.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/7/10.1182_bloodadvances.2018028456/2/m_advances028456f3.png?Expires=1770926912&Signature=g5pybpj~s-Bu7z778VjrYx5M~0YS6bJLC7Sf5zmeY2Lv4~9CVxJrlLqh1Ealj~7Rs9BFCG1FeLFHq~EeHK8DM9aBgDUH6hlHJAyZ88pFCDeaBMmFZMDfPQV6MykXUSm9OTQneOoi3XYGlp0P1wwI~hQ9hq9g5nkZYDLT-Pf7FCDjGLeyddFafaBOwM9RBmhpHuwiLiZGMcPYwEbog8~PvY6PDJ38EkTrJSLQifneKHT43gb2jRO2OS4lq379vFyPMw4UIrPeZsDazbXIbHXpFRo5bVvG~Hbbwb2TH1G5lNKgXmn-u~DRDuPFZ~J4tW9FCCqzhcZTx6EekYOWGj9UOw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CIT knockdown of MM cells reduced tumor growth in vivo. SCID/Beige mice were injected subcutaneously with OPM2 CIT-knockdown cells (n = 8) or scramble cells (n = 7) (5 × 106 per mouse) to establish an MM xenograft model. Tumor growth was measured, and volume was calculated as (length [mm] × width2 [mm2])/2. (A) CIT-knockdown cells showed reduced tumor growth in vivo (P < .001). (B) Mice bearing CIT-knockdown cells exhibited significantly prolonged survival (P < .001). (C) CIT expression of tumors collected from mice injected with scramble or CIT-knockdown cells. Decrease in CIT expression was confirmed ex vivo on harvested tumors using qRT-PCR. K.D, knockdown; Scr, scramble control.
CIT knockdown of MM cells reduced tumor growth in vivo. SCID/Beige mice were injected subcutaneously with OPM2 CIT-knockdown cells (n = 8) or scramble cells (n = 7) (5 × 106 per mouse) to establish an MM xenograft model. Tumor growth was measured, and volume was calculated as (length [mm] × width2 [mm2])/2. (A) CIT-knockdown cells showed reduced tumor growth in vivo (P < .001). (B) Mice bearing CIT-knockdown cells exhibited significantly prolonged survival (P < .001). (C) CIT expression of tumors collected from mice injected with scramble or CIT-knockdown cells. Decrease in CIT expression was confirmed ex vivo on harvested tumors using qRT-PCR. K.D, knockdown; Scr, scramble control.
TP53 expression level is an independent predictor of CIT dependency
Analysis of the Dependency Map’s genome-wide CRISPR screen data revealed that CIT is required for cell viability across all available MM cell lines, confirming our shRNA results and further establishing CIT as a potential therapeutic target for MM (Figure 4A). Because CIT dependency levels varied across MM cell lines, we applied linear regression to identify relevant biomarkers predicting strong CIT dependency. Variables that have been reported to interact or regulate CIT, including AURKA, AURKB, INCENP, CDK1, and TP53 gene expression, as well as TP53 mutation and copy number status, were included in the model. TP53 expression was an independent predictor of CIT dependency (multivariate P = 0.02), with low-TP53 cell lines exhibiting a strong dependency on CIT (Figure 4B).
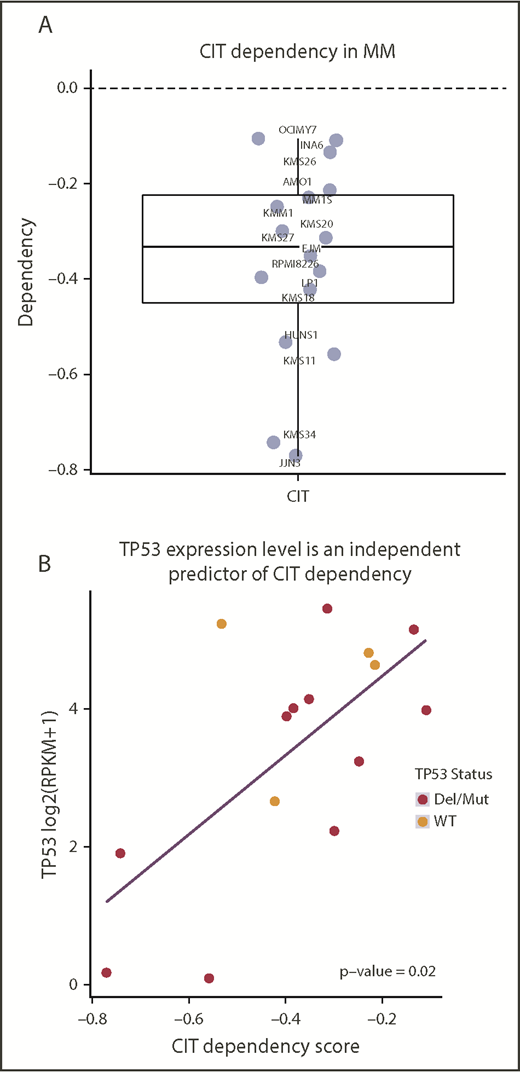
Dependency Map analysis. (A) CIT-dependency scores for available MM cell lines. (B) Multivariate analysis of CIT and related genes showing TP53 expression as an independent predictor of CIT dependency (multivariate P = .02).
Dependency Map analysis. (A) CIT-dependency scores for available MM cell lines. (B) Multivariate analysis of CIT and related genes showing TP53 expression as an independent predictor of CIT dependency (multivariate P = .02).
Discussion
CIT is highly enriched and plays a key role in the midbody, which regulates the final abscission of the 2 daughter cells at the end of cytokinesis.2,3 Despite its strong role in cytokinesis, the role of CIT in cancer pathogenesis is still lacking. Recent studies suggested that CIT is overexpressed in several carcinomas.5,6 Similarly, we demonstrated significantly higher CIT expression in MM plasma cells compared to healthy controls, which was confirmed by protein microarray, immunohistochemistry, and gene expression profiling. In contrast to solid tumor patients,6,16 MM patients with high CIT expression had significantly worse overall survival compared with patients with low CIT expression. Taken together, our data suggest that upregulation of CIT might have an important role in the pathogenesis of MM.
CIT messenger RNA was previously detected in adult murine tissues, including brain, kidney, spleen, thymus, skin, and lung but not liver, heart, or skeletal muscle.17 Strong CIT expression was found in highly proliferative cells, including testis and embryonic neuronal cells. Thus, it was thought that CIT’s expression could be cell cycle dependent rather than tissue specific, and low expression in some tissues may attributed to the quiescent state of those tissues.17 This property suggests that targeting CIT in tumor cells might result in less potential damage to the neighboring normal cells.
It is well known that CIT is a key regulator of the molecular network required for cytokinesis, and knockdown of this gene may disrupt cytokinesis and cell growth.2,3 Herein, we demonstrated that silencing CIT decreased MM cell proliferation in vitro, induced cytokinesis failure, and reduced tumor growth in vivo, which was also demonstrated previously in other tumor cell lines.5,18 Similar to our results, CIT upregulation was found in hepatocellular carcinoma, and its knockdown significantly inhibited hepatocellular carcinoma cell proliferation and in vivo tumorigenicity of the cells by interrupting cytokinesis.3 Another study suggested that citron kinase protein might be a potential target for medulloblastoma treatment.19 Cytokinesis failure can cause polyploidy, which is an irreversible process that prevents cell division and may lead to a reduction in tumor burden.20 In contrast, polyploidy can lead to chromosomal instability and increase tumorigenicity.21 These facts raise the interesting question of whether CIT silencing in hematologic malignancies may increase the risk of tumorigenicity of the cells while reducing tumor burden. It should also be noted that cytokinesis defects have been associated with induction of apoptosis,22 which could contribute to reduced tumor burden. The role and regulation of apoptosis in CIT knockdown in MM were not investigated in this study; further investigation into its mechanism of regulation in MM is warranted.
Interestingly, it has been shown that CIT may also have an important role in the G2/M transition in the cell cycle.4 In the current study, we observed a prolonged G2 arrest in CIT-knockdown OPM2 cells. However, this was not observed in CIT-knockdown MM.1s cells. We speculate that the use of different repair machinery systems by the 2 cell lines might partially explain that difference. Specifically, the MM.1s cell line has wild-type TP53, whereas the OPM2 cell line has mutant and deleted TP53.23 Intact p53 in MM.1S cells may partly explain the early arrest at the G1/S regulation point upon damage induction by CIT inhibition. In addition to that, a stronger antiproliferative effect was observed in the OPM2 cell line upon CIT inhibition. This result can also be explained by the different TP53 status between the OPM2 (mutant and deleted) and MM.1s (wild-type) cell lines, because we showed that TP53 expression is an independent predictor of CIT dependency, with low-TP53 cell lines being more sensitive to CIT inhibition.
Despite some initial reports that RhoA can activate CIT during cytokinesis,24,25 other reports suggest that CIT is required for correct RhoA localization at the cleavage site during late cytokinesis, indicating its role more as a RhoA regulator.26,27 It was also reported that CIT can contribute to the stabilization of midbody microtubules by indirectly altering the phosphorylation of tubulin β-III.28 The only known substrate of CIT is the inner centromere protein (INCENP), which is a component of the chromosomal passenger complex, and Aurora B is the kinase component of the chromosomal passenger complex. It should be noted that INCENP is also a target for Aurora B, which makes this interaction more complex.29 Recently, McKenzie et al30 demonstrated a cross talk between Aurora B and CIT. Their study showed that CIT can phosphorylate INCENP, which is required for aurora kinase activation. Reciprocally, Aurora B can phosphorylate CIT and control its localization. The role of aurora kinases was investigated in MM.31-33 Shi et al31 showed that targeting aurora kinases has antimyeloma effects, including the induction of tetraploidy followed by apoptosis. Further studies are needed to explore the interaction of 2 kinases in cancer pathogenesis, particularly in MM.
Given their significant role and function, primarily in cytokinesis, in contrast to other kinases (eg, Aurora B and Plk1) that have different roles throughout mitosis,34 we believe that small molecule inhibitors targeting CIT could be promising antiproliferative compounds in the treatment of MM. Moreover, we showed that cell lines with low TP53 expression due to mutations and deletions are particularly sensitive to CIT knockdown, which suggests a potential role for CIT inhibition in relapsed/refractory patients with biallelic TP53 inactivation. This is particularly important, because patients with biallelic TP53 inactivation exhibit significantly inferior outcomes.35 Nevertheless, it should be noted that cytokinesis failure could also promote tumorigenesis, as was observed in tetraploid cells devoid of p53.21 It has long been known that tumorigenesis can happen as a result of DNA-damaging therapy; however, melphalan, a DNA-damaging alkylator, is still among the most potent cytoreductors, with a well-established role in MM treatment. Although the role of CIT inhibition in MM treatment remains to be characterized, our results suggest great potential and lay the groundwork for drug development and further in vivo studies.
The full-text version of this article contains a data supplement.
Acknowledgment
This work was supported in part by a grant from the National Institutes of Health, National Cancer Institute (R01 CA181683).
Authorship
Contribution: I.S. performed research, analyzed data, and wrote the manuscript; Y.K., R.S.-P., M.M., Y.M., S.M., A.S., R.F., A.M.R., and T.W. performed research and analyzed data; R.C. performed immunohistochemistry; and I.M.G. designed the research and revised the manuscript.
Conflict-of-interest disclosure: R.F. has done consulting work for Celgene, Takeda, AbbVie, Amgen, Bayer, Janssen, Pharmacyclics, Merck, Sanofi, Kite, and Juno, and participated on a scientific advisory board for Adaptive Biotechnologies. The remaining authors declare no competing financial interests.
Correspondence: Irene M. Ghobrial, Department of Medical Oncology, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA 02115; e-mail: irene_ghobrial@dfci.harvard.edu.

