

Key Points
The CD1d-CD19 fusion binds to CD19+ targets and, once loaded with αGC, promotes iNKT cell activation.
The αGC-loaded CD1d-CD19 fusion induces robust iNKT cell lysis of CD19+ tumor cells in vitro and controls their growth in vivo.

Abstract
Invariant natural killer T (iNKT) cells comprise a unique lineage of CD1d-restricted lipid-reactive T lymphocytes that potently kill tumor cells and exhibit robust immunostimulatory functions. Optimal tumor-directed iNKT cell responses often require expression of the antigen-presenting molecule CD1d on tumors; however, many tumor cells downregulate CD1d and thus evade iNKT cell recognition. We generated a soluble bispecific fusion protein designed to direct iNKT cells to the site of B-cell cancers in a tumor antigen-specific but CD1d-independent manner. This fusion protein is composed of a human CD1d molecule joined to a single chain antibody FV fragment specific for CD19, an antigen widely expressed on B-cell cancers. The CD1d-CD19 fusion protein binds specifically to CD19-expressing, but not CD19-negative cells. Once loaded with the iNKT cell lipid agonist α-galactosyl ceramide (αGC), the CD1d-CD19 fusion induces robust in vitro activation of and cytokine production by human iNKT cells. iNKT cells stimulated by the αGC-loaded CD1d-CD19 fusion also strongly transactivate T-, B-, and NK-cell responses and promote dendritic cell maturation. Importantly, the αGC-loaded fusion induces robust lysis of CD19+CD1d− Epstein-Barr virus immortalized human B-lymphoblastoid cell lines that are otherwise resistant to iNKT cell killing. Consistent with these findings; administration of the αGC-loaded fusion protein controlled the growth of CD19+CD1d− tumors in vivo, suggesting that it can “link” iNKT cells and CD19+CD1d− targets in a therapeutically beneficial manner. Taken together, these preclinical studies demonstrate that this B cell–directed fusion protein can be used to effectively induce iNKT cell antitumor responses in vitro and in vivo.
Introduction
B-cell malignancies are among the most common cancers affecting children and adults and occur more frequently in patients with immunodeficiency.1 Approximately 10% to 20% of children and 50% of adults die from the disease. Unfortunately, recurrent or refractory B-cell malignancies are often resistant to standard therapies, with patients demonstrating poor tolerance of treatment because of organ dysfunction and/or increased susceptibility to infection.2,3 To improve the outcome for patients with advanced B-cell malignancies, we have developed an alternative therapeutic approach that capitalizes on the antitumor functions of invariant natural killer T (iNKT) cells.
iNKT cells are innate-type T lymphocytes, the majority of which express an “invariant” T-cell receptor (iTCR) that recognizes glycolipid antigens presented by the major histocompatibility complex (MHC) class I–like molecule CD1d.4,5 Following engagement of the iTCR by glycolipid/CD1d complexes, iNKT cells secrete cytokines, polarize cytolytic granules, and stimulate the functions of dendritic (DC), NK, T, and B cells. Based on their capacity to kill target cells and direct the development of innate and adaptive immune responses,6 several clinical trials have examined whether administration of specific glycolipids such as the prototypical iNKT agonist α-galactosyl ceramide (αGC), αGC-loaded DC or αGC-loaded DC and expanded human iNKT (hu-iNKT) cells might prove beneficial in the treatment of patients with cancer.7-12 Collectively, these studies have shown that iNKT cell therapies are well tolerated, increase the frequency of iNKT cells circulating in the blood and in tumor tissues, and induce a noticeable activation of the immune system.8,12 Despite these encouraging results, there is need for additional improvement. At present, the major challenges lie in better understanding how iNKT cells recognize and respond to various tumors and in developing new and improved methods to direct iNKT cells to the tumor site, where they can then exert their many functions.
Through prior studies, our study and that of others find that maximal iNKT cell killing requires the presentation of agonistic lipids such as αGC by CD1d on the tumor cells.13-15 However, tumor cells often downregulate CD1d and, as a result, become invisible to iNKT cell attack.16-19 To circumvent this critical barrier, we developed a bispecific fusion protein composed of a human CD1d molecule joined to a single chain antibody FV (scFV) fragment specific for CD19, an antigen expressed on the majority of human B-cell cancers. We hypothesized that this fusion would target iNKT cells to the site of B-cell cancers in a CD19-specific yet CD1d-independent manner. Indeed, our studies reveal that once loaded with αGC, the CD1d-CD19 fusion induces iNKT cell activation and enables iNKT cell killing of CD19+CD1d− tumor cells in vitro and controls their growth in vivo. Our studies indicate that the CD1d-CD19 fusion can link iNKT and tumor cells in a therapeutically relevant manner and provide a framework by which iNKT cell functions can be harnessed to treat relapsed or refractory CD19+ CD1d− B-cell cancers.
Materials and methods
Mice
C57BL/6 (B6) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed at the Children's Hospital of Philadelphia (Philadelphia, PA). The Institutional Animal Care and Use Committee at Children's Hospital of Philadelphia approved all experimental procedures.
Reagents
αGC (KRN7000) was purchased from Enzo Life Sciences (Farmingdale, NY) and the αGC analog PBS44 was a kind gift from Paul B. Savage (Brigham Young University, Provo, UT). To generate B16-CD19+ and MC38-CD19+ stable cell lines, murine B16 melanoma and MC38 colon carcinoma cells were transfected with a plasmid encoding human CD19 and cultured in the presence of G418 (Invitrogen, 1 mg/mL).
In vivo tumor model
B6 mice were engrafted subcutaneously with 7 × 105 MC38-CD19 cells and 72 hours later, injected IV every 2 to 3 days for a total of 6 doses with phosphate-buffered saline, αGC-loaded or unloaded CD1d-CD19 (40 μg), or αGC-loaded or unloaded “irrelevant” CD1d-Her2 scFV (40 μg). Tumor growth was measured using a digital caliper and tumor volume calculated using the formula: length × (width)2/2.
Statistics
Statistical analyses were performed using GraphPad PRISM software (San Diego, CA). P ≤ .05 was deemed significant.
For generation of the CD1d-CD19 fusion protein and details regarding in vitro assays, please refer to the supplemental Methods.
Results
Cloning, purification, and specificity of binding of the CD1d-CD19 fusion protein
To generate the CD1d-CD19 fusion protein, we fused a DNA fragment encoding human β2-microglobulin to the N terminus of human CD1d (Figure 1A-C; supplemental Figure 1). We then joined the C terminus of CD1d to an anti-human CD19 scFV, followed by a linker and 6-histidine tag. To evaluate the specificity of binding of αGC loaded (LFP) and unloaded versions (ULFP) of the fusion protein, we examined CD19+CD1d− Epstein-Barr virus immortalized human B-lymphoblastoid cell lines (EBV-LCL 1 and 2) cells, as well as CD1d– B16 and CD1d− MC38 cells (Figure 1D). We observed that the fusion protein did not bind to CD19−CD1d+ T cell lines (data not shown) or to native B16 or MC38 cells (Figure 1E). However, both the αGC-loaded and unloaded CD1d-CD19 fusion exhibited high levels of binding to B16 and MC38 cell lines stably transfected to express human CD19 as well as the CD19+ EBV-LCLs (Figure 1E). These data demonstrate that the CD19 targeting portion of the fusion is properly folded and retains specificity for binding to CD19+ but not CD19− target cells.
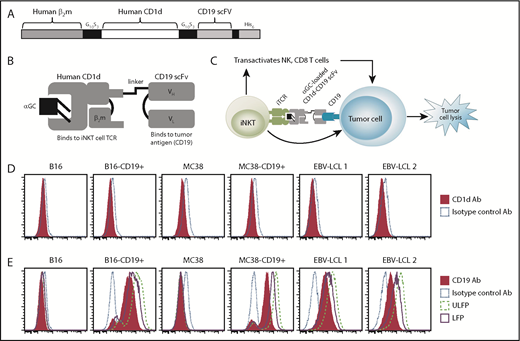
Design and binding specificity of the CD1d-CD19 fusion protein. (A) Schematic depicting the genetic fusion of human β2m with the sCD1d and the single chain of the human anti-CD19 antibody (CD19 scFV). DNA fragments were produced by polymerase chain reaction, including sequences for flexible glycine-serine rich linker (G10S3). A 6-histidine tag was added at the C terminus for Ni-NTA purification. (B) Illustration of the antigen-loaded fusion protein. (C) Depiction of how the CD1d-CD19 fusion “links” iNKT cells to tumor cells in a CD19-specific, yet CD1d-independent, manner (right). Once loaded with agonistic iNKT-directed lipids such as αGC, it is anticipated that the fusion will engage the iNKT TCR, induce iNKT activation, and promote lysis of CD19+CD1d− tumor cells (see visual abstract). (D) CD1d expression on the cell lines used in this study, as determined by staining target cells with a phycoerythrin-labeled isotype, control and anti-CD1d antibody. (E) Binding of the CD1d-CD19 fusion protein to CD1d− cell lines used in this study and detection of the fusion by a phycoerythrin-labeled anti-CD1d antibody. Data from >4 independent experiments are shown. Ab, antibody.
Design and binding specificity of the CD1d-CD19 fusion protein. (A) Schematic depicting the genetic fusion of human β2m with the sCD1d and the single chain of the human anti-CD19 antibody (CD19 scFV). DNA fragments were produced by polymerase chain reaction, including sequences for flexible glycine-serine rich linker (G10S3). A 6-histidine tag was added at the C terminus for Ni-NTA purification. (B) Illustration of the antigen-loaded fusion protein. (C) Depiction of how the CD1d-CD19 fusion “links” iNKT cells to tumor cells in a CD19-specific, yet CD1d-independent, manner (right). Once loaded with agonistic iNKT-directed lipids such as αGC, it is anticipated that the fusion will engage the iNKT TCR, induce iNKT activation, and promote lysis of CD19+CD1d− tumor cells (see visual abstract). (D) CD1d expression on the cell lines used in this study, as determined by staining target cells with a phycoerythrin-labeled isotype, control and anti-CD1d antibody. (E) Binding of the CD1d-CD19 fusion protein to CD1d− cell lines used in this study and detection of the fusion by a phycoerythrin-labeled anti-CD1d antibody. Data from >4 independent experiments are shown. Ab, antibody.
CD1d-CD19 fusion presents αGC to and activates hu-iNKT cells
To test whether the CD1d portion of the fusion can properly present αGC to iNKT cells, we cultured purified hu-iNKT cells on plates coated or not with varying concentrations of the unloaded or αGC-loaded fusion protein. For these studies, we used unfractionated iNKT cells consisting of 45.7 ± 2.2% CD4+, 4.6 ± 0.2% CD8+, and 46.3 ± 2.7% CD4−CD8− (double negative) iNKT cells (Figure 2A). As expected, incubation of iNKT cells with no or unloaded fusion failed to induce a response. In contrast, incubation with αGC-loaded fusion increased the expression of activation markers CD69 and CD25 with an average of two- to threefold increase in the mean fluorescence intensity. Responses were dose dependent and occurred at a concentration as low as 0.5 to 1.0 μg/mL (Figure 2B; data not shown). To assess whether the fusion promotes iNKT cell proliferation, experiments were repeated using iNKTs that had been labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) before placing into culture. As seen in Figure 2C, the αGC-loaded but not unloaded fusion protein promoted proliferation of iNKT cells, as demonstrated by the dilution of CFSE. Finally, analysis of culture supernatants revealed that the αGC-loaded fusion induced abundant secretion of numerous cytokines (Figure 2D). Thus, once loaded with an agonistic glycolipid antigen such as αGC, the CD1d component of the fusion can efficiently engage the iTCR and promote vigorous iNKT cell activation.
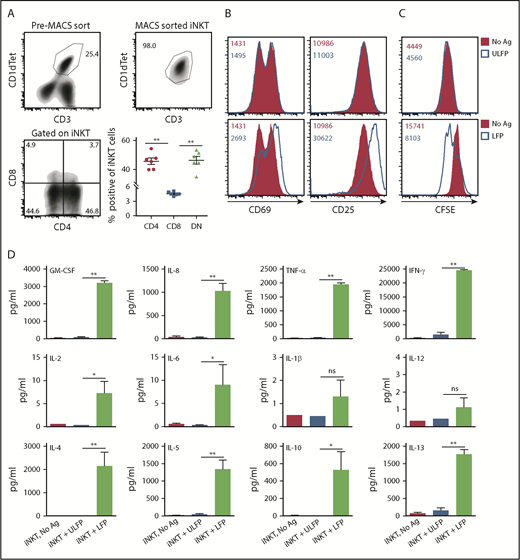
αGC-loaded CD1d-CD19 fusion promotes hu-iNKT cell activation in vitro. (A) hu-iNKT cells were expanded from PBMCs as detailed in “Materials and methods.” (Top left) Representative density plot showing % human CD1d tetramer (Cd1dtet)+CD3+ cells in the expanded PBMC cultures after 7 days. (Top right) Representative density plot showing that hu-iNKT cells can be successfully expanded from PBMCs and isolated to >98% purity. (Bottom left) Representative density plot showing percent CD4+ and percent CD8+ cells gated on iNKT cells from the PBMC cultures described previously. (Bottom right) Frequencies of CD4+, CD8+, and CD4−CD8− (DN) iNKT cells. Data are pooled from 6 independent donors. Each symbol in the graphs corresponds to 1 donor. (B) Freshly isolated hu-iNKT cells were cultured in plates coated with αGC-loaded or unloaded CD1d-CD19 fusion (1.0 μg/mL). After 24 hours, cells were harvested and analyzed for expression of activation markers (CD25 and CD69) by flow cytometry. (C) hu-iNKT cells were labeled with 250 nM of CFSE on day 0 and cultured as described. After 4 days, cells were harvested and analyzed for cell proliferation by flow cytometry. Numbers in the histograms indicate MFI. (D) Culture supernatants from experiments in panel A were analyzed for the levels of cytokines by MILLIPLEX MAP Human High Sensitivity T Cell Magnetic Bead Panel kit. Data are presented as mean ± standard error of the mean (SEM) and compiled from independent experiments using hu-iNKT cells from 10 normal donors. Significance was determined by unpaired 2-tailed Student t test. *P < .05, **P < .01. Ag, αGC; DN, double negative; GM-CSF, granulocyte-macrophage colony-stimulating factor; ns, not significant.
αGC-loaded CD1d-CD19 fusion promotes hu-iNKT cell activation in vitro. (A) hu-iNKT cells were expanded from PBMCs as detailed in “Materials and methods.” (Top left) Representative density plot showing % human CD1d tetramer (Cd1dtet)+CD3+ cells in the expanded PBMC cultures after 7 days. (Top right) Representative density plot showing that hu-iNKT cells can be successfully expanded from PBMCs and isolated to >98% purity. (Bottom left) Representative density plot showing percent CD4+ and percent CD8+ cells gated on iNKT cells from the PBMC cultures described previously. (Bottom right) Frequencies of CD4+, CD8+, and CD4−CD8− (DN) iNKT cells. Data are pooled from 6 independent donors. Each symbol in the graphs corresponds to 1 donor. (B) Freshly isolated hu-iNKT cells were cultured in plates coated with αGC-loaded or unloaded CD1d-CD19 fusion (1.0 μg/mL). After 24 hours, cells were harvested and analyzed for expression of activation markers (CD25 and CD69) by flow cytometry. (C) hu-iNKT cells were labeled with 250 nM of CFSE on day 0 and cultured as described. After 4 days, cells were harvested and analyzed for cell proliferation by flow cytometry. Numbers in the histograms indicate MFI. (D) Culture supernatants from experiments in panel A were analyzed for the levels of cytokines by MILLIPLEX MAP Human High Sensitivity T Cell Magnetic Bead Panel kit. Data are presented as mean ± standard error of the mean (SEM) and compiled from independent experiments using hu-iNKT cells from 10 normal donors. Significance was determined by unpaired 2-tailed Student t test. *P < .05, **P < .01. Ag, αGC; DN, double negative; GM-CSF, granulocyte-macrophage colony-stimulating factor; ns, not significant.
αGC-loaded CD1d-CD19 fusion promotes hu-iNKT cell-dependent transactivation of bystander immune cells
To investigate whether the αGC-loaded CD1d-CD19 fusion protein promotes iNKT cell-dependent DC maturation, we cultured peripheral blood mononuclear cells (PBMCs) with or without added purified autologous hu-iNKT cells on plates coated with unloaded or αGC-loaded CD1d-CD19 fusion protein. Twenty-four hours later, cells were harvested and analyzed by flow cytometry to assess the expression of costimulatory molecules on various antigen-presenting cell (APC) populations, such as such as CD19+ B cells or CD11c+ DC. In the absence of added autologous hu-iNKT, PBMC plated on unloaded or αGC-loaded fusion failed to upregulate HLA class II or the costimulatory molecules CD80 and CD86 on B cells or DCs (Figure 3A-B). On the other hand, coculture of PBMC with purified iNKT cells and immobilized αGC-loaded (but not unloaded) fusion dramatically increased the expression of these molecules on both APC populations (change in CD80 and CD86 mean fluorescence intensity [MFI] ranged from 2.5- to 4.2-fold increase; Figure 3A-B). Similarly, analysis of culture supernatants revealed that αGC-loaded (but not unloaded) fusion induced robust production of numerous cytokines, but only when iNKT was present (Figure 3C). When compared with the results of the prior experiments in which purified iNKT was plated on αGC-loaded fusion in the absence of added PBMC (Figure 2C), we observed that supernatants from cocultures containing PBMCs + iNKT cells exhibited much higher levels of interleukin-1β (IL-1β; produced by monocytes), IL-6 (produced by monocytes and macrophages), and IL-12 (produced by DC). In both sets of experiments, comparable numbers of iNKT were included and cells were stimulated for similar periods. Thus, these observations strongly suggest that these additional cytokines are not originating from iNKT cells themselves. Rather, the αGC-loaded fusion is activating iNKT cells, which in turn are driving cytokine production by other immune cells present in the cultures.
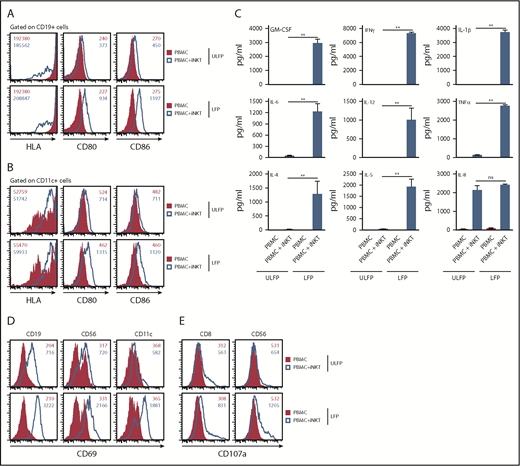
αGC-loaded CD1d-CD19 fusion promotes hu-iNKT cell-mediated transactivation of other immune cells in vitro. Freshly isolated hu-iNKT cells were mixed with autologous PBMCs at a 1:1 ratio (2 × 105 PBMCs: 2 × 105 NKT cells). Control wells had PBMCs only (4 × 105 cells). Cells were added to wells precoated with αGC-loaded or unloaded CD1d-CD19 fusion (1 μg/mL). After 24 hours, cells were harvested and analyzed for surface expression of HLA and costimulatory molecules on CD19+ B cells (A) and CD11c+ myeloid cells (B) by flow cytometry. (C) From the same experiments, culture supernatants were harvested and analyzed for cytokines as in Figure 2. Data are presented as mean ± SEM and compiled from independent experiments using autologous PBMCs and purified iNKT cells from 3 normal human donors. CD69 (D) and CD107a (E) expression was determined on different immune cell populations by flow cytometry. Data shown in panels A-B and D-E are representative histograms from 3 independent experiments. Numbers in the histograms indicate MFI. Significance was determined by unpaired 2-tailed Student t test. **P < .01.
αGC-loaded CD1d-CD19 fusion promotes hu-iNKT cell-mediated transactivation of other immune cells in vitro. Freshly isolated hu-iNKT cells were mixed with autologous PBMCs at a 1:1 ratio (2 × 105 PBMCs: 2 × 105 NKT cells). Control wells had PBMCs only (4 × 105 cells). Cells were added to wells precoated with αGC-loaded or unloaded CD1d-CD19 fusion (1 μg/mL). After 24 hours, cells were harvested and analyzed for surface expression of HLA and costimulatory molecules on CD19+ B cells (A) and CD11c+ myeloid cells (B) by flow cytometry. (C) From the same experiments, culture supernatants were harvested and analyzed for cytokines as in Figure 2. Data are presented as mean ± SEM and compiled from independent experiments using autologous PBMCs and purified iNKT cells from 3 normal human donors. CD69 (D) and CD107a (E) expression was determined on different immune cell populations by flow cytometry. Data shown in panels A-B and D-E are representative histograms from 3 independent experiments. Numbers in the histograms indicate MFI. Significance was determined by unpaired 2-tailed Student t test. **P < .01.
In the absence of added iNKTs, we observed no upregulation of CD69 on B (CD19+), NK (CD56+), or DCs (CD11c+) within the PBMC cultures, regardless of whether the cells had been plated on αGC-loaded or αGC-unloaded fusion protein (Figure 3D). Similarly, there was no upregulation of CD107a (LAMP1) on cytotoxic (CD8+) T or NK cells (Figure 3E). However, when cultures were supplemented with iNKT cells, all of these immune cell populations exhibited a dramatic increase in the surface expression of CD69 and CD107a (mean fold increase, 5.2- to 15.3-fold and 2.2- and 2.7-fold for CD69 and CD107a, respectively), especially in cultures plated on the αGC-loaded fusion. Of note, we did observe an increase in CD69 and CD107a expression, albeit to a lesser extent, when PBMC + iNKT were plated on unloaded fusion. Because our expanded iNKT cells are purified using an anti-iTCR antibody, it is possible that they become activated during this process. This iNKT cell activity may be sufficient to modestly stimulate other cells in the culture but insufficient to stimulate cytokine production (Figure 3C).
The αGC-loaded fusion induces hu-iNKT cell cytotoxic function
On closer analysis of the B-cell compartment in the PBMC+iNKT cocultures just described, we observed that there was a 72.1 ± 6.3% decrease in the incidence of CD19+ cells plated on αGC-loaded fusion. Strikingly, there was no change in the frequencies of CD4+, CD8+, CD56+, or CD11c+ cells (Figure 4A-B; data not shown) in the PBMC+iNKT cocultures plated on unloaded or αGC-loaded fusion protein. This decrease in the percentage of CD19+ cells is likely the result of cytolysis of B cells mediated by CD8+ or CD56+ cells (because they exhibit enhanced CD107a expression; Figure 3E) and/or via direct cytolysis by iNKT cells.
![Figure 4. αGC-loaded fusion promotes hu-iNKT cell cytotoxicity in vitro. (A-B) Freshly isolated hu-iNKT cells were mixed with autologous PBMCs and then added to wells pre-coated with αGC-loaded or unloaded CD1d-CD19 fusion as in Figure 3. After 24 hours, cells were harvested and analyzed for different immune cell populations by flow cytometry. (A) Representative density plots showing percent CD19+ B, percent CD4+, and percent CD8+ cells in PBMCs + iNKT cell mix after culture on plate-bound unloaded or loaded fusion protein (1 μg/mL) for 24 hours. (B) Frequencies of B (CD19+), CD4+, and CD8+ cells in PBMCs and iNKT cell cocultures plated on either unloaded or αGC-loaded CD1d-CD19 fusion protein. Data are pooled from 4 independent experiments. Each symbol in the graphs corresponds to 1 donor. (C) Freshly isolated human were plated with 51Cr-labeled EBV-LCLs at varying effector:target (E:T) ratios with no stimulus (no Ag), PBS44, or unloaded (ULFP) or αGC-loaded fusion (LFP, 1 μg/mL). After 16 to 18 hours, culture supernatants were collected, radioactivity quantified, and percent specific lysis calculated. Depicted are results obtained using 3 representative EBV-LCLs. Data are from 1 of 6 independent experiments. (D) Mean percent increase ± SEM in cytolysis of the EBV-LCLs in the presence of unloaded or αGC-loaded fusion compared with cytolysis in the presence of PBS44. Data are averaged from 6 EBV-LCL lines. (E) hu-iNKTs were plated with nonradiolabeled EBV-LCLs at varying E:T = 20:1 (40 × 103 NKT cells and 2 × 103 EBV-LCLs) ratio with no stimulus, or the unloaded or αGC-loaded CD1d-CD19 fusion (1 μg/mL). After 16 to 18 hours, supernatants were collected and IFN-γ levels were measured by enzyme-linked immunosorbent assay. Representative data (mean ± standard deviation [SD]) from 1 of 3 experiments is shown. (F) Data are as in panel D, except that in these experiments Jurkat (human, left) or EL4 (murine, right) T cells were used as targets. (G-H) Freshly isolated hu-iNKT cells were added to wells coated with plate-bound αGC-loaded or unloaded CD1d-CD19 fusion (1 μg/mL) or left untreated. (G) After 24 hours, cells were harvested and analyzed for intracellular levels of lytic molecule (granzyme B) and degranulation (CD107a). Data are representative of 3 independent experiments. Numbers in the histograms indicate MFI. (H) Compiled data (mean ± SD) from 3 independent experiments showing fold increase in granzyme B (GrB) and CD107a expression on iNKT cells plated on αGC-loaded CD1d-CD19 fusion compared with those plated on unloaded fusion. Significance in panels B, D, and H was determined by unpaired 2-tailed Student t test; and in panels C and F by 2-way analysis of variance (ANOVA) test. *P < .05, **P < .01.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/5/10.1182_bloodadvances.2018028886/2/m_advances028886f4.png?Expires=1768176415&Signature=32cPrvKUBSYqSdAexupJiYm-QQ6t28GgP2GidCMqKr~8CWVpFyeAlijshJVm2ZCJpQSdya9W0m~KblfpGeK2PAhHtcGW2hBD0xmndQ9yTQHfEYJEXTZyD2Cq2EkA9kjsyZjbwnSfAaGm16AikwOK4kqWmt0N1~cFbl-l5RPFjj~yue4IU9IntplZKl14uXFhHXRurM0uXKs-i2lThiCVLNs-MLko10ecneqCInSu5Amtp0TPy2qd~a3O14HBRUnkfOJUMW9EAhiWCc~A96asHHRLnpdu8~jV8c~xOGiC6zpQbitWDgGXXHKQkcBkkaVmEjGaZv30W9TUJERrM~ahBA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
αGC-loaded fusion promotes hu-iNKT cell cytotoxicity in vitro. (A-B) Freshly isolated hu-iNKT cells were mixed with autologous PBMCs and then added to wells pre-coated with αGC-loaded or unloaded CD1d-CD19 fusion as in Figure 3. After 24 hours, cells were harvested and analyzed for different immune cell populations by flow cytometry. (A) Representative density plots showing percent CD19+ B, percent CD4+, and percent CD8+ cells in PBMCs + iNKT cell mix after culture on plate-bound unloaded or loaded fusion protein (1 μg/mL) for 24 hours. (B) Frequencies of B (CD19+), CD4+, and CD8+ cells in PBMCs and iNKT cell cocultures plated on either unloaded or αGC-loaded CD1d-CD19 fusion protein. Data are pooled from 4 independent experiments. Each symbol in the graphs corresponds to 1 donor. (C) Freshly isolated human were plated with 51Cr-labeled EBV-LCLs at varying effector:target (E:T) ratios with no stimulus (no Ag), PBS44, or unloaded (ULFP) or αGC-loaded fusion (LFP, 1 μg/mL). After 16 to 18 hours, culture supernatants were collected, radioactivity quantified, and percent specific lysis calculated. Depicted are results obtained using 3 representative EBV-LCLs. Data are from 1 of 6 independent experiments. (D) Mean percent increase ± SEM in cytolysis of the EBV-LCLs in the presence of unloaded or αGC-loaded fusion compared with cytolysis in the presence of PBS44. Data are averaged from 6 EBV-LCL lines. (E) hu-iNKTs were plated with nonradiolabeled EBV-LCLs at varying E:T = 20:1 (40 × 103 NKT cells and 2 × 103 EBV-LCLs) ratio with no stimulus, or the unloaded or αGC-loaded CD1d-CD19 fusion (1 μg/mL). After 16 to 18 hours, supernatants were collected and IFN-γ levels were measured by enzyme-linked immunosorbent assay. Representative data (mean ± standard deviation [SD]) from 1 of 3 experiments is shown. (F) Data are as in panel D, except that in these experiments Jurkat (human, left) or EL4 (murine, right) T cells were used as targets. (G-H) Freshly isolated hu-iNKT cells were added to wells coated with plate-bound αGC-loaded or unloaded CD1d-CD19 fusion (1 μg/mL) or left untreated. (G) After 24 hours, cells were harvested and analyzed for intracellular levels of lytic molecule (granzyme B) and degranulation (CD107a). Data are representative of 3 independent experiments. Numbers in the histograms indicate MFI. (H) Compiled data (mean ± SD) from 3 independent experiments showing fold increase in granzyme B (GrB) and CD107a expression on iNKT cells plated on αGC-loaded CD1d-CD19 fusion compared with those plated on unloaded fusion. Significance in panels B, D, and H was determined by unpaired 2-tailed Student t test; and in panels C and F by 2-way analysis of variance (ANOVA) test. *P < .05, **P < .01.
αGC-loaded fusion promotes hu-iNKT cell cytotoxicity in vitro. (A-B) Freshly isolated hu-iNKT cells were mixed with autologous PBMCs and then added to wells pre-coated with αGC-loaded or unloaded CD1d-CD19 fusion as in Figure 3. After 24 hours, cells were harvested and analyzed for different immune cell populations by flow cytometry. (A) Representative density plots showing percent CD19+ B, percent CD4+, and percent CD8+ cells in PBMCs + iNKT cell mix after culture on plate-bound unloaded or loaded fusion protein (1 μg/mL) for 24 hours. (B) Frequencies of B (CD19+), CD4+, and CD8+ cells in PBMCs and iNKT cell cocultures plated on either unloaded or αGC-loaded CD1d-CD19 fusion protein. Data are pooled from 4 independent experiments. Each symbol in the graphs corresponds to 1 donor. (C) Freshly isolated human were plated with 51Cr-labeled EBV-LCLs at varying effector:target (E:T) ratios with no stimulus (no Ag), PBS44, or unloaded (ULFP) or αGC-loaded fusion (LFP, 1 μg/mL). After 16 to 18 hours, culture supernatants were collected, radioactivity quantified, and percent specific lysis calculated. Depicted are results obtained using 3 representative EBV-LCLs. Data are from 1 of 6 independent experiments. (D) Mean percent increase ± SEM in cytolysis of the EBV-LCLs in the presence of unloaded or αGC-loaded fusion compared with cytolysis in the presence of PBS44. Data are averaged from 6 EBV-LCL lines. (E) hu-iNKTs were plated with nonradiolabeled EBV-LCLs at varying E:T = 20:1 (40 × 103 NKT cells and 2 × 103 EBV-LCLs) ratio with no stimulus, or the unloaded or αGC-loaded CD1d-CD19 fusion (1 μg/mL). After 16 to 18 hours, supernatants were collected and IFN-γ levels were measured by enzyme-linked immunosorbent assay. Representative data (mean ± standard deviation [SD]) from 1 of 3 experiments is shown. (F) Data are as in panel D, except that in these experiments Jurkat (human, left) or EL4 (murine, right) T cells were used as targets. (G-H) Freshly isolated hu-iNKT cells were added to wells coated with plate-bound αGC-loaded or unloaded CD1d-CD19 fusion (1 μg/mL) or left untreated. (G) After 24 hours, cells were harvested and analyzed for intracellular levels of lytic molecule (granzyme B) and degranulation (CD107a). Data are representative of 3 independent experiments. Numbers in the histograms indicate MFI. (H) Compiled data (mean ± SD) from 3 independent experiments showing fold increase in granzyme B (GrB) and CD107a expression on iNKT cells plated on αGC-loaded CD1d-CD19 fusion compared with those plated on unloaded fusion. Significance in panels B, D, and H was determined by unpaired 2-tailed Student t test; and in panels C and F by 2-way analysis of variance (ANOVA) test. *P < .05, **P < .01.
To analyze whether the CD1d-CD19 fusion directly promotes hu-iNKT cell killing of CD19+ targets, we used the fusion in a 51Cr-release based killing assay in which EBV-LCL served as targets. We also examined the activation state of iNKT cells by culturing them with nonradio-labeled EBV-LCL and analyzed interferon-γ (IFN-γ) secretion. EBV-LCL express high levels of CD19 but lack CD1d (Figure 1B-C). As a result, EBV-LCL are not killed by iNKT cells, even in the presence of free PBS44 (structural analog of αGC) (Figure 4C). Strikingly, hu-iNKT cells exerted robust lysis of EBV-LCL (Figure 4C-D) and secreted increased levels of IFN-γ (Figure 4E) only when cultures contained αGC-loaded fusion. Thus, the CD1d-CD19 fusion targets iNKT cells to, and induces their functions at, the site of CD19+CD1d− B cells. Of note, hu-iNKT cells potently killed CD19−CD1d+ Jurkat (human) and CD19−CD1d+ EL4 (murine) T cells in the presence of free PBS44. However, the unloaded and the αGC-loaded CD1d-CD19 fusion failed to induce iNKT cell lysis of these CD1d+ T-cell lines (Figure 4F). These latter data suggest that once αGC is loaded onto the fusion, it remains bound and is not released into the medium where it can be then be loaded onto the CD1d+ T cells for presentation to the iNKT cells. Collectively, these findings demonstrate the specificity of the fusion protein and provide proof of principle that once loaded with an agonistic lipid such as αGC, the CD1d-CD19 fusion can link iNKT cells with CD19+CD1d− B-cell targets in a therapeutically relevant manner in vitro.
To determine whether the CD1d-CD19 fusion directly promotes iNKT cell degranulation or expression of death-inducing receptors, we cultured purified hu-iNKT cells in the presence of immobilized unloaded or αGC-loaded CD1d-CD19 fusion and measured the expression of the TRAIL and Fas ligand by flow cytometric analysis. We observed little to no difference in TRAIL and Fas ligand expression on hu-iNKT cells plated on either unloaded or αGC-loaded fusion (data not shown). In contrast, intracellular expression of granzyme B and CD107a in hu-iNKT cells significantly increased (2.85 ± 0.38-fold) in the presence of αGC-loaded fusion as compared with cells plated on unloaded fusion (Figure 4G-H). These data are consistent with studies implicating the perforin/granzyme B pathway as a mediator of iNKT cell killing.13,20
αGC-loaded fusion induces murine iNKT cell activation and immunomodulatory functions in vivo
Having demonstrated that the CD1d-CD19 fusion induces hu-iNKT cell activation in vitro, we next sought to determine whether it also induces iNKT cell functions in vivo. CD1d is highly conserved across species; we thus anticipated that the fusion would present αGC equally well to human or murine iNKTs. To examine this directly, we cultured sort-purified murine iNKT cells on plate-bound fusion protein. Consistent with the hu-iNKT data, αGC-loaded but not unloaded fusion increased murine iNKT cell CD69 and CD25 expression and induced robust IFN-γ production (supplemental Figure 2). To examine the kinetics of iNKT cell activation and further characterize the in vivo functional response induced by the αGC-loaded fusion, we injected wild-type B6 mice with a single dose (20 μg) of the unloaded or αGC-loaded fusion. At 2 and 18 hours postinjection, we harvested blood and organs and examined splenic and intrahepatic iNKT cell cytokine production (Figure 5A-C) and measured serum cytokine levels (Figure 5D). As reported previously,21 in vivo administration of PBS44 readily induced the production of IFN-γ by splenic and hepatic iNKT cells and this correlated with increased serum IFN-γ levels. In agreement with its inability to activate iNKT cells in vitro, the unloaded fusion failed to promote an in vivo iNKT cell response, even at the highest dose that was tested (40 μg; Figure 5; data not shown). In contrast, the αGC-loaded fusion induced activation of iNKT cells that in all cases mirrored the responses seen using free PBS44. Although the αGC-loaded fusion induced only modest intracellular IL-4 production by hepatic and splenic iNKT cells, IL-4 levels were undetectable in the serum (data not shown). These observations suggest that the αGC-loaded fusion promotes robust T helper 1 but not T helper 2 iNKT cell responses in vivo.

Antigen-loaded but not unloaded CD1d-CD19 fusion activates murine iNKT cell functions in vivo. B6 mice were injected intraperitoneally with PBS44 (4 μg), αGC-loaded or unloaded CD1d-CD19 fusion (20 μg), or left untreated. After 2 (A) or 18 (B) hours, splenic and hepatic iNKT cells producing IFN-γ (A-B) or IL-4 (C) directly ex vivo were analyzed by intracellular cytokine staining and flow cytometry. Data are representative of 3 experiments with 1 to 2 mice analyzed per condition. Numbers in the histograms indicate MFI. (D) Serum was collected at different time points as indicated on the graphs and IFN-γ levels were measured by enzyme-linked immunosorbent assay. Data represent the mean ± SEM from 3 independent experiments for each time point indicated. Significance was determined by unpaired 2-tailed Student t test. *P < .05, **P < .01.
Antigen-loaded but not unloaded CD1d-CD19 fusion activates murine iNKT cell functions in vivo. B6 mice were injected intraperitoneally with PBS44 (4 μg), αGC-loaded or unloaded CD1d-CD19 fusion (20 μg), or left untreated. After 2 (A) or 18 (B) hours, splenic and hepatic iNKT cells producing IFN-γ (A-B) or IL-4 (C) directly ex vivo were analyzed by intracellular cytokine staining and flow cytometry. Data are representative of 3 experiments with 1 to 2 mice analyzed per condition. Numbers in the histograms indicate MFI. (D) Serum was collected at different time points as indicated on the graphs and IFN-γ levels were measured by enzyme-linked immunosorbent assay. Data represent the mean ± SEM from 3 independent experiments for each time point indicated. Significance was determined by unpaired 2-tailed Student t test. *P < .05, **P < .01.
To examine whether the C1d-CD19 fusion enables activation of other immune cell lineages in vivo, we administered a single dose of free PBS44 or the unloaded or αGC-loaded fusion. After 18 hours, animals were euthanized and examined for upregulation of: (1) CD69 on splenic lymphocytes and myeloid cells (Figure 6A); (2) CD86 and MHCII on APCs (Figure 6B-C); and (3) IFN-γ production by NK cells (Figure 6D). In each of these assays, animals receiving free PBS44 or the αGC-loaded fusion exhibited comparable and robust responses, whereas those receiving unloaded fusion exhibited no response. Similar observations were made in the liver (data not shown). Taken together, these data demonstrate that the αGC-loaded fusion is fully functional and can induce systemic activation of the immune system in vivo.
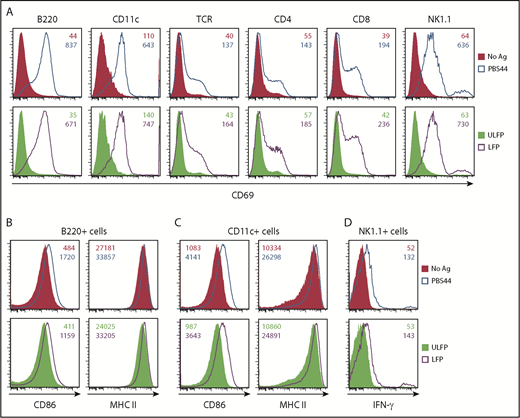
αGC-loaded but not the unloaded CD1d-CD19 fusion promotes murine iNKT cell immunomodulatory functions in vivo. B6 mice were injected intraperitoneally with PBS44 (4 μg), αGC-loaded or unloaded CD1d-CD19 fusion (20 μg), or left untreated. (A) After 18 hours, splenocytes were analyzed for TCR+, CD4+CD8+, NK1.1+, B220+, and CD11c+ cells expressing CD69. Surface expression of the costimulatory molecule CD86 and MHC II on splenic CD19+ B cells (B) and CD11c+ myeloid cells (C) was assessed by flow cytometry. (D) Splenocytes were stained with NK1.1 and TCRβ antibodies and the NK1.1+TCRβ− cells producing IFN-γ directly ex vivo were identified by intracellular staining and flow cytometry. Data are representative of 3 experiments with 1 to 2 mice analyzed per condition. Numbers in the histograms indicate MFI.
αGC-loaded but not the unloaded CD1d-CD19 fusion promotes murine iNKT cell immunomodulatory functions in vivo. B6 mice were injected intraperitoneally with PBS44 (4 μg), αGC-loaded or unloaded CD1d-CD19 fusion (20 μg), or left untreated. (A) After 18 hours, splenocytes were analyzed for TCR+, CD4+CD8+, NK1.1+, B220+, and CD11c+ cells expressing CD69. Surface expression of the costimulatory molecule CD86 and MHC II on splenic CD19+ B cells (B) and CD11c+ myeloid cells (C) was assessed by flow cytometry. (D) Splenocytes were stained with NK1.1 and TCRβ antibodies and the NK1.1+TCRβ− cells producing IFN-γ directly ex vivo were identified by intracellular staining and flow cytometry. Data are representative of 3 experiments with 1 to 2 mice analyzed per condition. Numbers in the histograms indicate MFI.
Treatment with αGC-loaded fusion protein controls tumor growth in vivo
To determine whether the αGC-loaded fusion is effective at controlling the growth of tumor cells in vivo, B6 mice were grafted with MC38 cells stably transfected to express human CD19 (MC38−CD19+). We used these cells for our studies because they lack CD1d expression (Figure 1B) and are amenable to growth in B6 mice. Additionally, MC38−CD19+ but not MC38 cells are susceptible to iNKT cell-mediated cytolysis in the presence αGC-loaded fusion (Figure 7A; data not shown). Three days posttumor cell injection; mice were treated with no fusion protein (group I) or equivalent amounts (40 μg) of αGC-loaded or unloaded CD1d-CD19 fusion (groups II and III), or with an “irrelevant” αGC-loaded or unloaded CD1d-Her2 fusion protein (groups IV and V). Except for the untreated animals (group I), mice received a total of 6 injections of fusion protein over a 3-week course. Consistent with our in vitro and in vivo data, mice treated with the unloaded CD1d-CD19 fusion were unable to inhibit tumor growth (group III, Figure 7), similar to animals that received no fusion protein (group I, Figure 7). In contrast, we observed maximal control of tumor growth following administration of αGC-loaded CD1d-CD19 fusion protein (group II, Figure 7). Mice in this group either had very small tumors <300 mm3 or had no visible or palpable tumors. The requirement for tumor-targeting (ie, CD19-directed) effects was best demonstrated in the mice treated with the irrelevant (Her2-directed) antigen-loaded fusion (group IV, Figure 7) because all of the animals in this group had large tumors, indistinguishable from animals receiving no treatment (group I, Figure 7) or treatment with unloaded fusion (groups III and V, Figure 7). MC38−CD19+ tumor-bearing mice treated with repeated injections of free αGC (total of 5 IV injections over 3 weeks) failed to control tumor growth, similar to mice treated with unloaded fusion (data not shown). Altogether, these data identify the CD1d-CD19 fusion as a novel tool with which to effectively direct the antitumor functions of iNKT cells to the site of B-leukemia/lymphoma cells in vitro and in vivo.
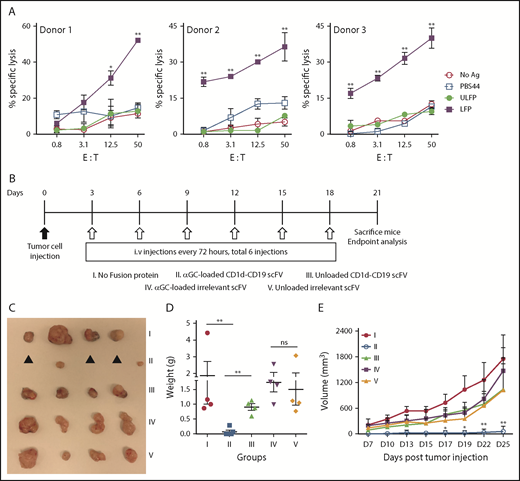
Treatment with the αGC-loaded CD1d-CD19 fusion limits tumor growth in vivo. (A) Freshly isolated hu-iNKT cells were plated with 51Cr-labeled MC38-CD19+ at varying E:T ratios with no stimulus (no Ag), PBS44, or ULFP or LFP (1 μg/mL). After 16 to 18 hours, culture supernatants were collected, radioactivity quantified, and percent specific lysis calculated. Depicted are results obtained using iNKT cells from 3 independent healthy donors. (B) Mice were grafted subcutaneously with 700 000 MC38-CD19+ cells, and treatment was started 72 hours later. Mice were treated every 3 days for a total of 6 doses CD1d-CD19 with either phosphate-buffered saline (control), αGC-loaded or unloaded CD1d-CD19 fusion (40 μg), or equivalent amounts of αGC-loaded or unloaded irrelevant Cd1d-Her2 fusion (40 μg). Mice were analyzed after 3 weeks. (C) Excised tumors from 1 representative experiment. ▲, no visible or palpable tumors. Weight (D) and volume (E) of excised tumors. Data in panels D-E are presented as mean ± SD of 4 to 5 mice per group from 1 of 2 independent experiments. Significance was determined by unpaired 2-tailed Student t test (D) or 2-way ANOVA (A,E). *P < .05, **P < .01.
Treatment with the αGC-loaded CD1d-CD19 fusion limits tumor growth in vivo. (A) Freshly isolated hu-iNKT cells were plated with 51Cr-labeled MC38-CD19+ at varying E:T ratios with no stimulus (no Ag), PBS44, or ULFP or LFP (1 μg/mL). After 16 to 18 hours, culture supernatants were collected, radioactivity quantified, and percent specific lysis calculated. Depicted are results obtained using iNKT cells from 3 independent healthy donors. (B) Mice were grafted subcutaneously with 700 000 MC38-CD19+ cells, and treatment was started 72 hours later. Mice were treated every 3 days for a total of 6 doses CD1d-CD19 with either phosphate-buffered saline (control), αGC-loaded or unloaded CD1d-CD19 fusion (40 μg), or equivalent amounts of αGC-loaded or unloaded irrelevant Cd1d-Her2 fusion (40 μg). Mice were analyzed after 3 weeks. (C) Excised tumors from 1 representative experiment. ▲, no visible or palpable tumors. Weight (D) and volume (E) of excised tumors. Data in panels D-E are presented as mean ± SD of 4 to 5 mice per group from 1 of 2 independent experiments. Significance was determined by unpaired 2-tailed Student t test (D) or 2-way ANOVA (A,E). *P < .05, **P < .01.
Discussion
In this study, we generated and tested the antitumor activities of a bispecific fusion protein comprising a human CD1d molecule fused to an antibody single chain variable fragment specific for the human B-cell antigen CD19. Once loaded with the lipid agonist αGC, the CD1d-CD19 fusion promoted robust iNKT cell activation, proliferation, and cytokine production, and enabled secondary iNKT cell-dependent activation of T, B, NK, and myeloid cells. Importantly, it facilitated iNKT cell lysis of CD1d−CD19+ B-leukemia cells in vitro and controlled the growth of CD1d−CD19+ tumor cells in vivo. These studies demonstrate the tremendous translational potential of the CD1d-CD19 fusion as a novel means to harness the antitumor activities of iNKT cells as a treatment of refractory or relapsed CD1d−CD19+ B-cell malignancies.
This study highlights several important findings that are relevant to use of the CD1d-CD19 fusion as a novel anticancer agent. For example, iNKT cells activated via the αGC-loaded fusion produce high levels of cytokines, most notably IFN-γ (Figure 2C). Production of IFN-γ by iNKT cells promotes antitumor immune responses by stimulating the functions of cytotoxic lymphocytes.22-24 Consistent with this notion, we observe robust activation of CD8+ T and NK cells (upregulation of activation markers and evidence of degranulation) when iNKT cells are cocultured with PBMC and αGC-loaded fusion (Figure 3D-E). Interestingly, we find that the αGC-loaded fusion also induces production of IL-10 by iNKT cells (Figure 2C). A recent report identifies a role for IL-10 in the activation and expansion of iNKT cells.25 Thus, it is possible that iNKT cell–derived IL-10 acts in a positive feedback loop to boost iNKT cell responses.
Once activated, iNKT cells induce the maturation of DC, which then drive the activation of other immune cells.26-28 In these studies, IL-12 levels were markedly increased in supernatants from cocultures containing hu-iNKT, PBMC, and αGC-loaded CD1d-CD19 fusion (Figure 3C). In contrast, there was little to no IL-12 in the supernatants from cultures containing only hu-iNKT and the αGC-loaded fusion (Figure 2C). These data suggest that the increased IL-12 in the iNKT/PBMC cocultures is likely derived from activated DCs (and possibly also macrophages), which serve as the primary sources of this cytokine. Consistent with this notion, the DC in these cocultures exhibited marked upregulation of the activation marker CD69 and the costimulatory molecules CD80 and CD86 (Figure 3B,D). In addition to IL-12, we observed significantly elevated levels of IL-1β and IL-6, cytokines produced by monocytes and macrophages, in cocultures containing iNKT, PBMC, and αGC-loaded CD1d-CD19 fusion (Figure 3C). Thus, the αGC-loaded fusion serves as a potent stimulus for DC and other myeloid-lineage cells, as well as iNKT cells.
We and others have shown that human and murine iNKT cells mount robust CD1d-dependent cytotoxic responses against T- and B-lymphoblastic leukemia cells.13,17,21,29-31 In these prior studies, CD1d expression on tumor cells was essential to enable iNKT-mediated lysis. Indeed, it has been demonstrated that downregulation of CD1d is 1 means by which a B-cell lymphoma can evade the host antitumor immune response.14,16-18 Similarly, transformation of B cells by EBV results in the complete loss of CD1d expression and the inability to activate iNKT cells even in the presence of exogenous αGC.32 Consistently, we observed that CD1d−CD19+ EBV-LCL and CD1d−CD19+ MC38 cells were resistant to killing by hu-iNKT cells (Figures 4C-D and 7A). However, in the presence of the αGC-loaded fusion, iNKT cells mediated rapid and robust lysis of these cells. These data highlight the relevance of using the αGC-loaded CD1d-CD19 fusion to bring iNKT cells to the site of B-cell targets, even if these targets have low or no surface CD1d expression.
We find that iNKT cells kill CD1d+CD19− T-lymphoblastic leukemia cells in the presence of free exogenously added αGC analog PBS44 (Figure 4F), but fail to do so in the presence of the αGC-loaded fusion. This finding highlights 2 important concepts. First, once loaded onto the fusion, αGC is not released into the medium, where it can then be presented by CD1d+ cells to stimulate iNKT cells. Second, the activation of iNKT cells by the αGC-loaded fusion protein is tumor antigen-directed and restricted by the specificity of the scFV portion of the fusion. Consistent with this second point, the frequencies of B (but not DC or T or NK) cells were reduced in cocultures of containing iNKT, PBMC, and αGC-loaded fusion (Figure 4A-B; data not shown). Mechanistically, we observe that the αGC-loaded fusion induced granzyme and CD107 expression on hu-iNKT (Figure 4G-H; data not shown), in agreement with prior studies demonstrating that iNKT cells primarily mediate their cytotoxic effects via the exocytosis of perforin-containing lytic granules.13,20
Importantly, we observe that the αGC-loaded but not unloaded fusion mediates robust activation of iNKT cells as well as other cytotoxic lymphocytes in vivo (Figures 5 and 6), where its administration effectively controlled the growth of CD1d−CD19+ MC38 tumor cells (Figure 7B-E). In contrast, administration of free αGC (data not shown), unloaded fusion or an αGC-loaded fusion with an irrelevant scFV portion completely failed to restrain tumor growth. These findings underscore the importance of the antigen specificity of the fusion (in this case, targeted toward CD19) in mediating control of tumor growth, which likely results from the recruitment of iNKT and other immune cells to, and activation of these cells locally at, the site of CD19-expressing tumors. Indeed, a prior study testing the in vivo effects of a Her2-directed fusion reports that it can induce the localization and accumulation of iNKT, NK, and T cells at the site of Her2+ tumors.33
In conclusion, these studies provide a strong rationale for further development of CD1d fusions such as the one reported here as a tool in the fight against human cancers, particularly those that lack CD1d expression. Circulating iNKT cell numbers are diminished in patients with myeloid and lymphoid leukemias, B-cell non-Hodgkin lymphoma and multiple myeloma.34-39 In many of these patients, iNKT cells also exhibit functional defects.36,37 Herein, we demonstrate that the αGC-loaded CD1d-CD19 fusion induces robust proliferation of iNKT and other immune cells, but only in the presence of target cells expressing CD19. Accordingly, this fusion could be used to induce iNKT cell expansion and activation in vivo similar to bispecific T-cell engaging antibodies such as blinatumomab,40 or it could be used to expand iNKT cells in vitro for subsequent adoptive cellular therapy. Invariant NKT cells enhance antitumor responses by recalibrating exhausted T cells41 and increase the efficacy of anticancer vaccines.42 Therefore, in addition to its use as a single agent, the αGC-loaded CD1d-CD19 fusion could be used in combination with other cell-based therapies to nucleate and sustain effective immune responses against relapsed or refractory B-cell cancers.
The full-text version of the article contains a data supplement.
Acknowledgments
The authors thank the National Institutes of Health Tetramer Core Facility for generating PBS57-loaded CD1d tetramers.
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (R21 AI113490) (K.E.N.) and National Cancer Institute (5K22CA188149-02) (R.D.), Vaccinex (K.E.N.), Cookies for Kids Cancer (K.E.N.), and the SAS Foundation for Cancer Research (R.D.).
Authorship
Contribution: R.D. designed and carried out experiments, interpreted data, and wrote and edited the manuscript; P.G., T.G.G., S.J.W., and N.P.P. provided technical assistance; E.E. and M.Z. provided the murine B16 melanoma and MC38 colon carcinoma cell lines as well as the various antigen-loaded and unloaded fusion protein; and K.E.N oversaw the project, interpreted data, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for S.J.W. is UPMC Children's Hospital of Pittsburgh, Pittsburgh, PA.
The current affiliation for N.P.P. is Department of Obstetrics and Gynecology, Newark Beth Israel Medical Center, Newark, NJ.
Correspondence: Kim E. Nichols, St. Jude Children’s Research Hospital, 262 Danny Thomas Blvd, Memphis, TN 38105; e-mail: kim.nichols@stjude.org; and Rupali Das, Michigan State University, 567 Wilson Rd, BPS 2195, East Lansing, MI 48824; e-mail: dasrupal@msu.edu.

