

Key Points
The heat shock protein 90 inhibitor LAM-003 displays potent in vitro and in vivo activity as a single agent and combined with venetoclax.
LAM-003 retains antileukemic activity against AML cells rendered resistant to FLT3 kinase inhibitors by mutation or stromal signaling.

Abstract
Acute myeloid leukemias (AML) harboring a constitutively active internal tandem duplication (ITD) mutation in the FMS-like kinase tyrosine kinase (FLT3) receptor are associated with poor patient prognosis. Despite initial clinical responses to FLT3 kinase inhibitors, patients eventually relapse. Mechanisms of resistance include the acquisition of secondary FLT3 mutations and protective stromal signaling within the bone marrow niche. Here we show that LAM-003, a prodrug of the heat shock protein 90 inhibitor LAM-003A, has cytotoxic activity against AML cell lines and primary samples harboring FLT3-ITD. LAM-003 regressed tumors in an MV-4-11 xenograft mouse model and extended survival in a MOLM-13 systemic model. LAM-003 displayed synergistic activity with chemotherapeutic drugs and FLT3 inhibitors, with the most robust synergy being obtained with venetoclax, a BCL-2 inhibitor. This finding was verified in a MOLM-13 systemic survival model in which the combination significantly prolonged survival compared with the single agents. Importantly, LAM-003 exhibited equipotent activity against FLT3 inhibitor–resistant mutants of FLT3, such as D835 or F691, in cytotoxic and FLT3 degradation assays. LAM-003 also retained potency in AML cells grown in stromal-conditioned media that were resistant to FLT3 inhibitors. Lastly, a genome-wide CRISPR screen revealed epigenetic regulators, including KDM6A, as determinants of LAM-003 sensitivity in AML cell lines, leading to the discovery of synergy with an EZH2 inhibitor. Collectively, these preclinical findings support the use of LAM-003 in FLT3-ITD patients with AML who no longer respond to FLT3 inhibitor therapy either as a single agent or in combination with drugs known to be active in AML.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by the proliferation and accumulation of myeloid cells in the bone marrow. Overexpression of FMS-like kinase tyrosine kinase 3 (FLT3) receptor occurs in nearly all cases of AML,1 and mutations in FLT3 represent one of the most common genetic alterations, occurring in ∼30% of patients.2 Approximately 75% of these are internal tandem duplication (ITD) mutations, and 25% are point mutations in the activation loop of the tyrosine kinase domain (TKD), consisting mostly of D835 mutations.3 These mutations are activating, and the presence of an FLT3-ITD mutation confers poor prognosis.4-6 Two FLT3 tyrosine kinase inhibitors (FLT3i), midostaurin and gilteritinib, have been approved for treatment; not all patients respond, however, and those who do inevitably relapse due to resistance from acquisition of secondary mutations in FLT3,7,8 upregulation of other molecular pathways,9 or influence of the bone marrow microenvironment.10-14 As such, novel therapies that can overcome these resistance mechanisms are needed for patients harboring FLT3-ITD that are refractory to, or no longer respond to, therapy with FLT3i.
Heat shock protein 90 (HSP90) is an adenosine triphosphate–dependent chaperone required for the stabilization of client proteins. In cancer cells, HSP90 stabilizes oncoproteins that are often overexpressed or mutated.15 HSP90 has recently been shown to be a hub for a highly integrated complex of proteins (epichaperome) in tumor cells that enhances cell survival.16
FLT3 and FLT3 harboring the ITD mutation are client proteins of HSP90 and subject to degradation by HSP90 inhibitors (HSP90i).17-19 As such, HSP90 inhibition in cell lines or primary AML blasts results in cell death.17-22 Furthermore, HSP90i display greater potency toward 32D murine bone marrow cells harboring various FLT3 mutations, including TKD mutations that confer resistance to FLT3i, compared with cells expressing wild-type FLT3 (WT FLT3).19 These observations are further supported by the findings from several unbiased screening efforts in which libraries of 122,23 160,24 187,21 and 34925 anticancer drugs showed that HSP90i were among the most active drugs, especially against AML cells harboring FLT3-ITD, with little effect observed on cells from healthy donors. Detailed analyses correlating drug response to gene expression and mutations identified a significant association between HSP90i and AML cells harboring the FLT3-ITD mutation.23,24 Collectively, these findings suggest that inhibiting HSP90 is an effective strategy to target AML cells harboring FLT3-ITD and may be efficacious in patients relapsed or refractory to FLT3i. Although HSP90i have undergone extensive evaluation in the clinic,26 only 3 trials have targeted AML27-29 and reported limited clinical activity (18% at best).27 However, no trial has focused on the genetically defined FLT3-ITD patient population.
LAM-003A (formerly MPC-310030 ) is an orally bioavailable HSP90i that was well tolerated in a phase 1 trial in patients with relapsed or refractory solid tumors.31 The recommended phase 2 dose was determined to be 480 mg/d. However, the poor solubility of LAM-003A necessitated that it be formulated as a large tablet containing 40% Captisol (CyDex Pharmaceuticals, Inc.). To improve the formulation, an l-alanine ester prodrug, LAM-003, was synthesized.32 Compared with LAM-003A, LAM-003 displays excellent aqueous solubility and undergoes rapid bioconversion to the parent drug without the need for solubilizing excipients, resulting in a small tablet dosage form.
Here we report our preclinical studies of LAM-003 focusing on FLT3-ITD AML. We show that LAM-003 displayed significant antileukemic activity in vitro and in vivo in AML driven by FLT3-ITD and that LAM-003 was synergistic with the BCL-2 inhibitor venetoclax. LAM-003 also overcame the resistance to FLT3i conferred by stromal conditions or secondary mutations in FLT3. Furthermore, a genome-wide CRISPR screen uncovered genes that determine sensitivity to LAM-003. We validated the top hit KDM6A, a histone H3K27 demethylase. Accordingly, inhibition of EZH2, the histone H3K27 methyltransferase that functionally opposes KDM6A, synergized with LAM-003. Together, these data form the preclinical rationale for testing LAM-003 in the first clinical trial to evaluate the use of an HSP90i, either alone or in combination, in AML patients with a genetically defined mutation, specifically those harboring FLT3-ITD whose disease has progressed on FLT3i-based regimens (NCT03426605).
Methods
Cell culture
AML cell lines were obtained from DSMZ or ATCC and cultured according to instructions. Primary AML samples were obtained from Yale University, Weill Cornell Medicine, and NewYork-Presbyterian Hospital from patients with written informed consent and according to a protocol approved by the University Investigation Committees or purchased from commercial vendors. Specific details are provided in the supplemental Methods and materials.
In vivo studies
All in vivo studies were performed under approved animal care protocols and the standards of the Association for Assessment and Accreditation of Laboratory Animal Care. Specific details of each study performed are provided in the supplemental Methods and materials.
Western blot analysis
Western blot analysis was performed as previously described.33 Antibody details are provided in the supplemental Methods and materials.
Cell viability
AML cell lines, primary AML samples, or BA/F3 cells were treated with indicated drugs for 72 hours. Cell viability was measured by using Annexin V and 7-AAD staining (BioLegend) or the CellTiter-Glo Luminescent Cell Viability reagent (Promega) according to the manufacturer’s instructions. Further details are provided in the supplemental Methods and materials.
Drug combination studies
The combination index values were computed by using the Chou-Talalay method.34 Details of all drug combination studies are provided in the supplemental Methods and materials.
CRISPR screen
Statistical analysis
GraphPad Prism Version 6 (GraphPad Software, Inc.) was used for Kaplan-Meier analysis of animal survival, Student t test analysis, and analysis of variance. The P values are indicated in the figure legends and represented by asterisks in each figure according to P < .05 (*), P < .01 (**), P < .001 (***) and P < .0001 (****). Specific details are provided in the supplemental Methods and materials.
Results
LAM-003 displays single-agent activity and synergizes with antileukemic drugs in AML cells harboring FLT3-ITD
The prodrug LAM-003 is rapidly metabolized by esterases in vivo to the parent drug, LAM-003A.32 The bioconversion to parent drug also occurs in cell culture medium, in which ∼90% conversion occurs within 24 hours (supplemental Figure 1A). We confirmed that LAM-003 conversion in vitro generates the biologically active LAM-003A as MV-4-11 cells treated with LAM-003 for 24 hours induced HSP70 expression, a well-established pharmacodynamic biomarker of HSP90 inhibition,36 in a dose-dependent manner (supplemental Figure 1B).
The antileukemic activity of LAM-003 was tested in AML cell lines and primary blasts harboring WT FLT3 or FLT3-ITD/D835 mutations. We noted that cells harboring FLT3-ITD and/or D835 mutations exhibited greater sensitivity to LAM-003 than those with WT FLT3 mutations (Figure 1A; supplemental Figure 1C). LAM-003 also reduced colony formation ability more effectively in cell lines harboring FLT3-ITD (supplemental Figure 1D). Thus, AML cells harboring FLT3-ITD represent a genetic subset that is particularly sensitive to LAM-003. Given the especially poor prognosis for patients with FLT3-ITD AML, we focused our study on this subset. We confirmed that LAM-003 degrades FLT3 in cell lines harboring FLT3-ITD. MV-4-11 and MOLM-13 cells treated with LAM-003 for 24 hours showed >65% reduction in FLT3 cell surface expression as assessed by using flow cytometry (Figure 1B). Consistently, we observed reductions in pAKT(S473), pSTAT5(Y694), and pSyk(Y352)/pZAP-70(Y319) as well as pS6(S235/236), a robust readout for oncogenic FLT3-ITD signaling.37
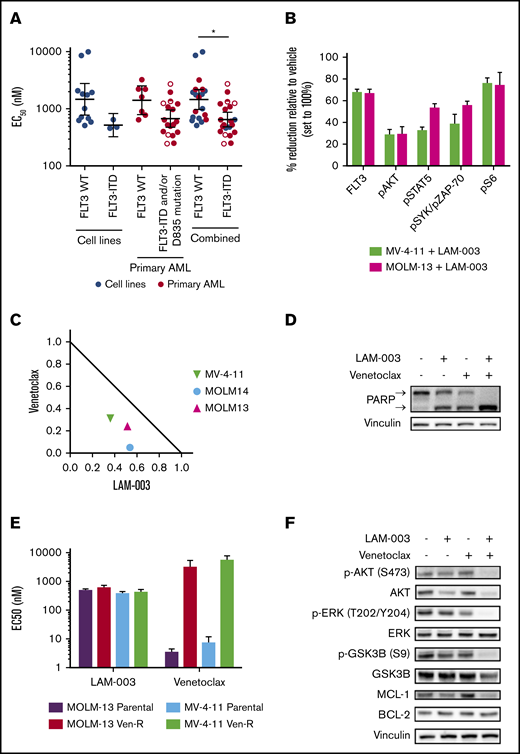
LAM-003 display antileukemic activity in AML cells harboring FLT3-ITD and synergistic activity with venetoclax. (A) Dot plot of average EC50 values in AML cell lines harboring WT FLT3 (WT) (n = 12) or FLT3-ITD (n = 3) or primary blasts (FLT3 WT, n = 7; FLT3-ITD and/or D835 mutation, n = 18) treated with LAM-003 for 72 hours. Cell lines were tested in duplicate a minimum of 2 independent times, and primary samples were tested once, in duplicate. The geometric mean ± 95% confidence interval is shown. Open circles are primary samples harboring D835 mutations, half circles are primary samples harboring FLT3-ITD and D835 mutation. EC50 values were calculated as described in the supplemental Methods and materials and shown in supplemental Table 1. *P < .05 using unpaired Student t test with Welch’s correction. (B) Expression of FLT3 and phospho-S6, phospho-AKT, phospho-STAT5, and phospho-SYK/ZAP-70 was evaluated by using flow cytometry in MV-4-11 (green bars) and MOLM-13 (pink bars) cells after treatment with LAM-003 for 24 hours. Average data ± standard deviation (SD) from 2 independent experiments are shown. (C) Normalized isobologram at the EC75 of three FLT3-ITD–harboring cell lines treated with a combination of LAM-003 and venetoclax for 72 hours before viability was assayed by using CellTiter-Glo. Each data point is the average of 2 independent experiments, each performed in duplicate. (D) Western blot analysis of MOLM-14 cells treated with LAM-003 (1 µM), venetoclax (20 nM), or the combination for 24 hours. Lysates were probed with antibodies to PARP or vinculin, which was used as a loading control. Upper and lower arrows denote full-length PARP and cleaved PARP, respectively. Representative data shown from 2 independent experiments. (E) EC50 values of LAM-003 or venetoclax in parental MOLM-13 or MV-4-11 cells and venetoclax-resistant cell lines (Ven-R) treated for 72 hours before viability determined by using CellTiter-Glo luminescent cell viability reagent. Experiments were performed a minimum of 2 independent times, each in duplicate, and averaged data ± SD are shown. (F) Western blot analysis of MOLM-14 cells treated with LAM-003 (1 µM), venetoclax (20 nM), or the combination for 24 hours. Lysates were probed with the indicated antibodies. Vinculin was used as a loading control. Representative blot shown from 2 independent experiments.
LAM-003 display antileukemic activity in AML cells harboring FLT3-ITD and synergistic activity with venetoclax. (A) Dot plot of average EC50 values in AML cell lines harboring WT FLT3 (WT) (n = 12) or FLT3-ITD (n = 3) or primary blasts (FLT3 WT, n = 7; FLT3-ITD and/or D835 mutation, n = 18) treated with LAM-003 for 72 hours. Cell lines were tested in duplicate a minimum of 2 independent times, and primary samples were tested once, in duplicate. The geometric mean ± 95% confidence interval is shown. Open circles are primary samples harboring D835 mutations, half circles are primary samples harboring FLT3-ITD and D835 mutation. EC50 values were calculated as described in the supplemental Methods and materials and shown in supplemental Table 1. *P < .05 using unpaired Student t test with Welch’s correction. (B) Expression of FLT3 and phospho-S6, phospho-AKT, phospho-STAT5, and phospho-SYK/ZAP-70 was evaluated by using flow cytometry in MV-4-11 (green bars) and MOLM-13 (pink bars) cells after treatment with LAM-003 for 24 hours. Average data ± standard deviation (SD) from 2 independent experiments are shown. (C) Normalized isobologram at the EC75 of three FLT3-ITD–harboring cell lines treated with a combination of LAM-003 and venetoclax for 72 hours before viability was assayed by using CellTiter-Glo. Each data point is the average of 2 independent experiments, each performed in duplicate. (D) Western blot analysis of MOLM-14 cells treated with LAM-003 (1 µM), venetoclax (20 nM), or the combination for 24 hours. Lysates were probed with antibodies to PARP or vinculin, which was used as a loading control. Upper and lower arrows denote full-length PARP and cleaved PARP, respectively. Representative data shown from 2 independent experiments. (E) EC50 values of LAM-003 or venetoclax in parental MOLM-13 or MV-4-11 cells and venetoclax-resistant cell lines (Ven-R) treated for 72 hours before viability determined by using CellTiter-Glo luminescent cell viability reagent. Experiments were performed a minimum of 2 independent times, each in duplicate, and averaged data ± SD are shown. (F) Western blot analysis of MOLM-14 cells treated with LAM-003 (1 µM), venetoclax (20 nM), or the combination for 24 hours. Lysates were probed with the indicated antibodies. Vinculin was used as a loading control. Representative blot shown from 2 independent experiments.
Having shown LAM-003 single-agent cytotoxic activity, we determined whether LAM-003 displayed synergistic activity with clinical stage drugs (cytarabine, daunorubicin, sorafenib, crenolanib, gilteritinib, or venetoclax). The most robust response was observed with venetoclax, the selective BCL-2 inhibitor, where synergy occurred in all three FLT3-ITD AML cell lines (Figure 1C) with the lowest average combination index34 values (Table 1). Synergy between LAM-003 and venetoclax was corroborated by assessing PARP integrity as a marker of apoptosis, which exhibited complete cleavage upon combinatorial drug treatment compared with either drug alone (Figure 1D; supplemental Figure 2A).
Summary of combination index values in three FLT3-ITD AML cell lines treated with LAM-003 in combination with those drugs used for AML
| Cell line | |||
|---|---|---|---|
| Combination LAM-003 + | MV-4-11 | MOLM-13 | MOLM-14 |
| Daunorubicin | 0.6 | 0.9 | 0.8 |
| Cytarabine | 0.8 | 1.0 | 0.9 |
| Crenolanib | 0.7 | 0.9 | 1.0 |
| Sorafenib | 0.8 | 1.0 | 1.0 |
| Gilteritinib | 1.0 | 0.9 | 1.0 |
| Venetoclax | 0.7 | 0.3 | 0.6 |
| Cell line | |||
|---|---|---|---|
| Combination LAM-003 + | MV-4-11 | MOLM-13 | MOLM-14 |
| Daunorubicin | 0.6 | 0.9 | 0.8 |
| Cytarabine | 0.8 | 1.0 | 0.9 |
| Crenolanib | 0.7 | 0.9 | 1.0 |
| Sorafenib | 0.8 | 1.0 | 1.0 |
| Gilteritinib | 1.0 | 0.9 | 1.0 |
| Venetoclax | 0.7 | 0.3 | 0.6 |
We next evaluated whether LAM-003 and venetoclax synergy depended on basal BCL-2 levels. We confirmed that venetoclax sensitivity correlated with BCL-2 levels in 9 of 10 of the cell lines tested (supplemental Figure 2B), as expected.38 Notably, synergy was observed in all 7 cell lines with high BCL-2 levels, and no synergy was observed in cell lines with low BCL-2 levels. Interestingly, all 3 (100%) FLT3-ITD cell lines had high BCL-2 levels, whereas only 4 (57%) of the 7 WT FLT3 cell lines tested had high BCL-2 levels.
To explore the mechanism underlying LAM-003 and venetoclax synergy, we focused on MCL-1 because its increased abundance confers resistance to venetoclax.39,40 We assayed LAM-003 in venetoclax-resistant FLT3-ITD AML cell clones due to overexpression of MCL-140 and found that LAM-003 retained equipotent antileukemic activity compared with parental cells (Figure 1E). Furthermore, cotreatment with LAM-003 and venetoclax markedly reduced MCL-1 (Figure 1F; supplemental Figure 2C-D). We next investigated the status of AKT, an upstream kinase that regulates the degradation of MCL-1 through inhibitory phosphorylation of GSK3β(Ser 9),41 and found that the combination of LAM-003 and venetoclax also resulted in loss of pAKT(S473), degradation of AKT, and subsequent loss of GSK3β(Ser 9) phosphorylation. Furthermore, MCL-1 degradation and PARP cleavage (supplemental Figure 2E-F) and synergy (supplemental Figure 2G) were also observed in the MV-4-11 and MOLM-13 venetoclax-resistant cell lines and in WT FLT3 cell lines (supplemental Figure 2H) after the combination treatment. These findings predicted that inhibiting AKT would phenocopy the effects of LAM-003 and synergize with venetoclax. Indeed, MOLM-13, MOLM-14, and MV-4-11 cells showed synergistic cell death when treated with venetoclax and the AKT inhibitors MK-2206 or GSK690693 (supplemental Figure 2I). Finally, we confirmed that pharmacological inhibition of MCL-1 and BCL-2 is synergistic in FLT3-ITD AML cell lines (supplemental Figure 2J) as previously observed,42 consistent with our mechanistic data. These findings suggest that the combination of LAM-003 and venetoclax confers synergy by destabilizing MCL-1 through AKT degradation (supplemental Figure 2K).
LAM-003 exhibits potent in vivo antileukemic activity as a single agent and in combination
To translate the in vitro studies, the efficacy of LAM-003 was tested by using MV-4-11 cells in an AML xenograft model. Leukemia-bearing animals were randomized into 2 groups and dosed orally once a day with either vehicle or LAM-003. LAM-003 exhibited potent antitumor activity, with an overall 84% tumor regression and no significant effect on body weight (supplemental Figure 3A-B). Importantly, the drug exposure at this efficacious 150 mg/kg dose (AUC24h = 104 917 ng h/mL) (supplemental Figure 3C) is clinically achievable based on phase 1 data with the parent drug, LAM-003A.31 To assess whether LAM-003 could improve the survival of animals with systemic AML, the MOLM-13 model was used. Animals dosed with LAM-003 (75 or 150 mg/kg) orally once a day exhibited a dose-dependent statistically significant improvement in survival compared with animals dosed with vehicle (Figure 2A). We also evaluated the single-agent activity of LAM-003 in the venetoclax-resistant MOLM-13 in vivo systemic model.40 Animals dosed with LAM-003 (125 mg/kg) orally once a day exhibited reduced AML burden in the peripheral blood after 13 days of treatment and had a significant increase in survival compared with animals dosed with vehicle (supplemental Figure 3D-E).
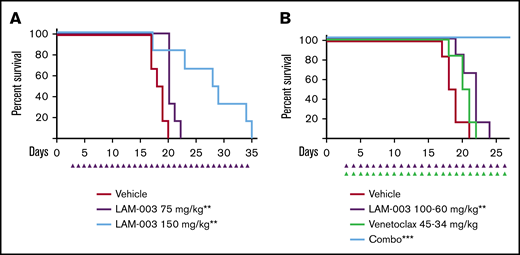
LAM-003 exhibits potent antileukemic activity in AML mouse models. (A) Kaplan-Meier survival analysis of a MOLM-13 systemic model in which mice were dosed orally with vehicle or with LAM-003 (75 or 150 mg/kg daily). Statistical significance was calculated by using the log-rank (Mantel-Cox) test. **P = .005 for LAM-003 75 mg/kg vs vehicle; **P = .008 for LAM-003 150 mg/kg vs vehicle. (B) Kaplan-Meier analysis of animal survival of mice inoculated with MOLM-13 cells systemically and treated with LAM-003 orally, venetoclax orally, or the combination as indicated once daily. Statistical significance was calculated by using the log-rank (Mantel-Cox) test. **P = .008 for LAM-003 vs vehicle; ***P = .0006 for the drug combination vs vehicle.
LAM-003 exhibits potent antileukemic activity in AML mouse models. (A) Kaplan-Meier survival analysis of a MOLM-13 systemic model in which mice were dosed orally with vehicle or with LAM-003 (75 or 150 mg/kg daily). Statistical significance was calculated by using the log-rank (Mantel-Cox) test. **P = .005 for LAM-003 75 mg/kg vs vehicle; **P = .008 for LAM-003 150 mg/kg vs vehicle. (B) Kaplan-Meier analysis of animal survival of mice inoculated with MOLM-13 cells systemically and treated with LAM-003 orally, venetoclax orally, or the combination as indicated once daily. Statistical significance was calculated by using the log-rank (Mantel-Cox) test. **P = .008 for LAM-003 vs vehicle; ***P = .0006 for the drug combination vs vehicle.
Finally, based on the in vitro data showing the synergistic activity of LAM-003 and venetoclax in FLT3-ITD AML cells, and the single-agent activity in vivo, the combination of LAM-003 and venetoclax was evaluated in the MOLM-13 systemic model. Leukemia-bearing animals were treated with suboptimal doses of LAM-003, venetoclax, or the combination. Although there was no significant improvement in survival with single-agent venetoclax, LAM-003 significantly improved survival compared with vehicle. Importantly, the combination of LAM-003 and venetoclax induced a dramatic effect on survival as 100% of the animals survived to the experimental end point (Figure 2B). These findings show that the combination of LAM-003 and venetoclax is highly efficacious in vivo.
LAM-003 overcomes mutations that confer resistance to FLT3i
To explore the clinical utility of LAM-003 in FLT3i relapsed/refractory FLT3-ITD AML, we evaluated its efficacy under conditions that confer resistance to FLT3i. Secondary mutations in FLT3 are a key intrinsic mechanism conferring resistance to FLT3i.11 We therefore tested whether LAM-003 was effective in degrading FLT3-harboring secondary mutations by introducing WT FLT3 or the following mutants into BA/F3 cells: FLT3-ITD, FLT3 D835V, FLT3-ITD D835V, FLT3 D835Y, FLT3-ITD D835Y, FLT3 D835H, FLT3-ITD D835H, FLT3 F691L, and FLT3-ITD F691L. Assaying these engineered lines, LAM-003 dose dependently reduced FLT3 mutant cell surface expression, which was found to be ∼5 times more sensitive than WT FLT3 (Figure 3A; Table 2). Moreover, LAM-003 displayed greater potency against FLT3i-resistant point mutations independently of FLT3 status, consistent with a requirement for HSP90 to stabilize the mutant proteins. To confirm whether these effects translated to increased antiproliferative activity, LAM-003 was tested against the FLT3-ITD F691L gatekeeper mutation, which confers resistance to crenolanib.37 Indeed, the FLT3-ITD F691L mutation induced a 23-fold decrease in crenolanib potency compared with cells harboring FLT3-ITD (Figure 3B). In contrast, LAM-003 displayed similar antileukemic activity in both FLT3-ITD mutant cell lines (FLT3-ITD half maximal effective dose [EC50] = 497 nM; FLT3-ITD-691L EC50 = 391 nM). To determine whether LAM-003 was also efficacious against human FLT3-ITD AML cells resistant to FLT3i, a cell line harboring a D835Y mutation was tested. The D835 mutation is a clinically relevant mutation as it has been shown to emerge in patients progressing while on sorafenib treatment.7 Parental MOLM-13 and MOLM-13 cells harboring FLT3-ITD and D835Y (MOLM-13-RES D835Y) mutations were treated with increasing concentrations of sorafenib, tandutinib, or LAM-003 for 72 hours. MOLM-13-RES D835Y cells displayed the expected resistance to tandutinib and sorafenib37 compared with parental cells (∼50 and 300 times, respectively). In contrast, LAM-003 was equipotent in the FLT3i-resistant and parental cell lines (Figure 3C).
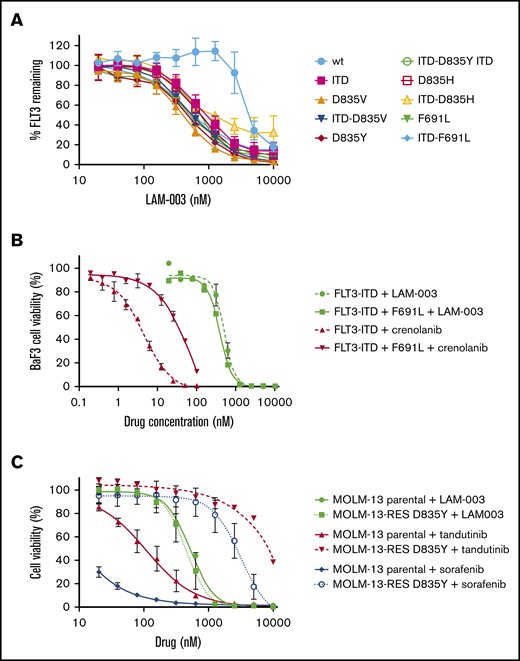
LAM-003 overcomes mutations that confer FLT3i resistance. (A) Ba/F3 cells harboring WT FLT or indicated FLT3 mutants were treated with increasing concentrations of LAM-003 for 24 hours before flow cytometry was performed to assess FLT3 expression. (B) Ba/F3 cells expressing FLT3-ITD or FLT3-ITD F691L were treated with crenolanib or LAM-003 for 72 hours. (C) Parental MOLM-13 cells (solid lines) or MOLM13-RES cells (hatched lines) were treated with tandutinib, sorafenib, or LAM-003 at the indicated concentrations for 72 hours before viability was assayed by using CellTiter-Glo. Data in all panels represent the mean ± SD from at least 2 independent experiments, each performed in duplicate.
LAM-003 overcomes mutations that confer FLT3i resistance. (A) Ba/F3 cells harboring WT FLT or indicated FLT3 mutants were treated with increasing concentrations of LAM-003 for 24 hours before flow cytometry was performed to assess FLT3 expression. (B) Ba/F3 cells expressing FLT3-ITD or FLT3-ITD F691L were treated with crenolanib or LAM-003 for 72 hours. (C) Parental MOLM-13 cells (solid lines) or MOLM13-RES cells (hatched lines) were treated with tandutinib, sorafenib, or LAM-003 at the indicated concentrations for 72 hours before viability was assayed by using CellTiter-Glo. Data in all panels represent the mean ± SD from at least 2 independent experiments, each performed in duplicate.
EC50 values of LAM-003–induced degradation of WT FLT3 and FLT3 mutants in BA/F3 cells
| Client protein | EC50, nM |
|---|---|
| WT FLT3 | 3590 |
| FLT3-ITD | 751 |
| FLT3 D835V | 406 |
| FLT3-ITD D835V | 545 |
| FLT3 D835Y | 509 |
| FLT3-ITD D835Y | 504 |
| FLT3 D835H | 662 |
| FLT3-ITD D835H | 440 |
| FLT3 F691L | 662 |
| FLT3-ITD F691L | 448 |
| Client protein | EC50, nM |
|---|---|
| WT FLT3 | 3590 |
| FLT3-ITD | 751 |
| FLT3 D835V | 406 |
| FLT3-ITD D835V | 545 |
| FLT3 D835Y | 509 |
| FLT3-ITD D835Y | 504 |
| FLT3 D835H | 662 |
| FLT3-ITD D835H | 440 |
| FLT3 F691L | 662 |
| FLT3-ITD F691L | 448 |
LAM-003 overcomes extrinsic factors that confer resistance to FLT3i
Because the bone marrow provides an environment that confers resistance to FLT3i,12,13 this may limit durable effects. As such, we tested the efficacy of LAM-003 under several experimental conditions. Bone marrow stromal factors can be replicated with human stromal cell–conditioned medium.13 FLT3-ITD–harboring cell lines were treated with gilteritinib, crenolanib, or LAM-003 for 72 hours in either nonconditioned or conditioned medium. In accordance with previous reports, all cell lines demonstrated significantly increased resistance to gilteritinib and crenolanib when grown in conditioned medium compared with nonconditioned medium (Figure 4A). Notably, LAM-003 retained potency in cells treated in conditioned medium compared with nonconditioned medium, at clinically achievable concentrations. In addition, MOLM-13-luc+ cells when coincubated with HS-5 stromal cells exhibited significant resistance to midostaurin, as expected,43 yet LAM-003 remained equally active under the 2 conditions (supplemental Figure 4).
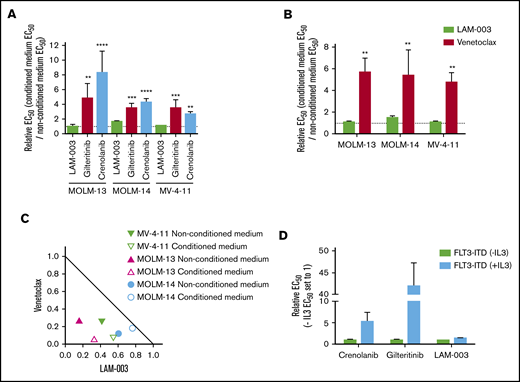
LAM-003 overcomes intrinsic FLT3 TKI resistance mechanisms. (A) AML cell lines were treated with LAM-003, gilteritinib, or crenolanib for 72 hours in either conditioned medium or nonconditioned medium before cell viability was determined by using CTG. All comparisons were made to LAM-003 by using a 1-way analysis of variance, Dunnett’s multiple comparisons test. For MOLM-13, **P = .007, ****P < .0001; for MOLM-14, ***P = .0002, ****P < .0001; for MV-4-11, **P = .002, ****P < .0001. (B) AML cell lines were treated with LAM-003 or venetoclax for 72 hours in either conditioned medium or nonconditioned medium. For MOLM-13, **P = .001; for MOLM-14, **P < .005; for MV-4-11, **P < .007 compared with LAM-003 using 2-way analysis of variance, Bonferroni’s multiple comparisons test. (C) Normalized isobologram at the EC75 of MV-4-11 cells and MOLM-13 cells with the combination of LAM-003 and venetoclax in nonconditioned (solid symbol) or conditioned (open symbol) medium for 72 hours before viability was assayed by using CellTiter-Glo. Each data point is the average of 2 independent experiments, each performed in duplicate, for each cell line. (D) EC50 values of BA/F3 cells expressing WT-FLT3 or FLT3-ITD supplemented with or without IL-3 and treated with gilteritinib, crenolanib, or LAM-003. Viability was assessed 72 hours later by using CTG. Data in all panels are the mean of at least 2 independent experiments, each performed in duplicate. Bar graphs in panels A-B and D represent the mean ± SD.
LAM-003 overcomes intrinsic FLT3 TKI resistance mechanisms. (A) AML cell lines were treated with LAM-003, gilteritinib, or crenolanib for 72 hours in either conditioned medium or nonconditioned medium before cell viability was determined by using CTG. All comparisons were made to LAM-003 by using a 1-way analysis of variance, Dunnett’s multiple comparisons test. For MOLM-13, **P = .007, ****P < .0001; for MOLM-14, ***P = .0002, ****P < .0001; for MV-4-11, **P = .002, ****P < .0001. (B) AML cell lines were treated with LAM-003 or venetoclax for 72 hours in either conditioned medium or nonconditioned medium. For MOLM-13, **P = .001; for MOLM-14, **P < .005; for MV-4-11, **P < .007 compared with LAM-003 using 2-way analysis of variance, Bonferroni’s multiple comparisons test. (C) Normalized isobologram at the EC75 of MV-4-11 cells and MOLM-13 cells with the combination of LAM-003 and venetoclax in nonconditioned (solid symbol) or conditioned (open symbol) medium for 72 hours before viability was assayed by using CellTiter-Glo. Each data point is the average of 2 independent experiments, each performed in duplicate, for each cell line. (D) EC50 values of BA/F3 cells expressing WT-FLT3 or FLT3-ITD supplemented with or without IL-3 and treated with gilteritinib, crenolanib, or LAM-003. Viability was assessed 72 hours later by using CTG. Data in all panels are the mean of at least 2 independent experiments, each performed in duplicate. Bar graphs in panels A-B and D represent the mean ± SD.
Stromal signaling also confers resistance to venetoclax,13 suggesting one explanation for the limited clinical activity of venetoclax as a single agent in patients with relapsed and refractory AML.44 We confirmed that although conditioned medium induced venetoclax resistance in FLT3-ITD cell lines, LAM-003 remained equipotent under the 2 conditions (Figure 4B). Moreover, the combination of LAM-003 and venetoclax remained synergistic in conditioned medium in the FLT3-ITD cell lines (Figure 4C). Finally, we used the BA/F3 cell line (interleukin 3 [IL-3]–dependent) to show that introduction of FLT3-ITD is sufficient for IL-3–independent survival yet renders the cells sensitive to the FLT3i gilteritinib and crenolanib. However, resistance to FLT3i is conferred by alternative pathway activation through addition of IL-3, as expected.45 In contrast, LAM-003 retains equal antileukemic activity (Figure 4D). Collectively, these findings show that in contrast to FLT3i, LAM-003 maintains potency under alternative pathway activation.
LAM-003 activity against FLT3i-resistant FLT3-ITD AML primary blasts
We determined the activity of LAM-003 against primary human AML blasts harboring FLT3-ITD. Primary blasts exhibited a dose-dependent increase in Annexin V/7-AAD staining after treatment with LAM-003 for 72 hours (Figure 5A). Importantly, one sample (Y1265) was obtained from a patient who had refractory FLT3-ITD AML progressing after cytarabine/idarubicin induction chemotherapy and high-dose chemotherapy/total body irradiation with hematopoietic progenitor cell transplantation, and who had relapsed after gilteritinib therapy. The blasts were confirmed to be resistant to gilteritinib (EC50 > 100 nM) but exhibited a dose-dependent reduction in viability upon treatment with LAM-003 (extrapolated EC50 = 505 nM) (Figure 5B; supplemental Table 1). We next tested whether the combination of LAM-003 and venetoclax was synergistic in 2 primary AML samples, both harboring FLT3-ITD. Primary AML cells were treated for 72 hours before viability was assayed by Annexin V/7-AAD staining on CD45+/CD34+ AML blasts. Figure 5C shows that LAM-003 and venetoclax treatment of primary AML also resulted in synergistic cell death.
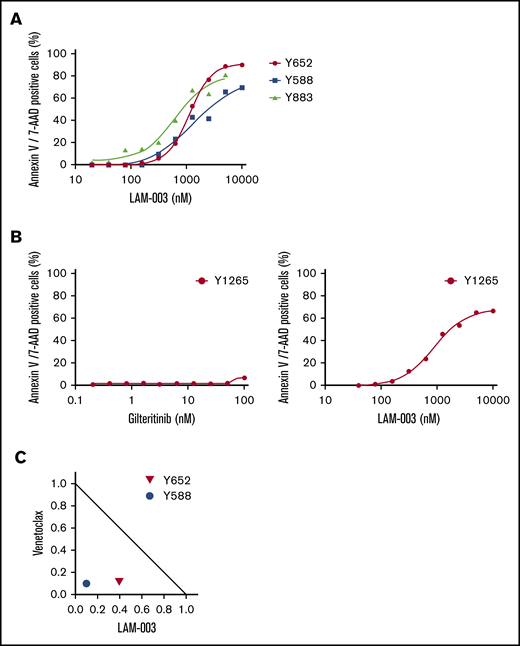
LAM-003 activity against TKI-resistant primary AML blasts harboring FLT3-ITD. (A) Three primary AML samples all harboring FLT3-ITD were treated with LAM-003 for 72 hours before Annexin V/7-AAD staining was assessed on CD45+/CD34+ blasts. (B) Primary AML blasts from a TKI-relapsed patient were treated with gilteritinib (left) or LAM-003 (right) for 72 hours before cell viability was assessed by using 7-AAD/Annexin V staining as in panel A. (C) Normalized isobologram at the EC75 of 2 primary AML samples, both harboring FLT3-ITD, with the combination of LAM-003 and venetoclax for 72 hours before being assayed for Annexin V/7-AAD staining as in panel A.
LAM-003 activity against TKI-resistant primary AML blasts harboring FLT3-ITD. (A) Three primary AML samples all harboring FLT3-ITD were treated with LAM-003 for 72 hours before Annexin V/7-AAD staining was assessed on CD45+/CD34+ blasts. (B) Primary AML blasts from a TKI-relapsed patient were treated with gilteritinib (left) or LAM-003 (right) for 72 hours before cell viability was assessed by using 7-AAD/Annexin V staining as in panel A. (C) Normalized isobologram at the EC75 of 2 primary AML samples, both harboring FLT3-ITD, with the combination of LAM-003 and venetoclax for 72 hours before being assayed for Annexin V/7-AAD staining as in panel A.
Genome-wide CRISPR screen identifies epigenetic regulation as a determinant of LAM-003 sensitivity
To identify genes that confer resistance to LAM-003 upon deletion, we conducted a CRISPR-mediated, genome-wide loss-of-function screen in the MOLM-14 cell line grown in the presence of 1 µM LAM-003. The GeCKO V2 library46 was used to perform this genetic screen as outlined in supplemental Figure 5. Genomic DNA harvested from surviving cells was analyzed for the identification of enriched single-guide RNAs (sgRNAs) across both GeCKO sublibraries (Figure 6A). Gene ontology analysis of the top 20 enriched hits across both GeCKO sublibraries identified epigenetic regulation, chromatin organization, and chromatin-modifying enzymes as the most highly enriched pathways in the pools surviving LAM-003 treatment (Figure 6B). The most enriched gene from the screen was KDM6A, a histone H3K27 demethylase47 (Figure 6C). Loss-of-function mutations of KDM6A are observed in FLT3-ITD AML,48 and such mutations or knock out of KDM6A confer resistance to chemotherapy in AML,49 confirming the clinical relevance of further exploring its role in regulating LAM-003 sensitivity. CRISPR-mediated targeting of the KDM6A locus was validated at the DNA, RNA, and functional levels (supplemental Methods and materials; supplemental Figure 6A-D). Pertinently, we observed that 3 independent sgRNAs targeting KDM6A conferred resistance to LAM-003 in the MOLM-14 and MV4-11 cell lines (supplemental Figure 6E). To therapeutically exploit this finding, we hypothesized that inhibiting EZH2, a histone-lysine N-methyltransferase that functionally opposes KDM6A, would enhance sensitivity to LAM-003. Two FLT3-ITD cell lines were tested with the combination of LAM-003 and a clinical stage EZH2 inhibitor (EPZ-6438), and synergy was observed in both cell lines (Figure 6D). These findings show that epigenetic regulators can influence LAM-003 sensitivity, that loss-of-function mutations in such genes may be useful as biomarkers of LAM-003 activity, and that targeting epigenetic regulators may be combined with LAM-003 for therapeutic use.
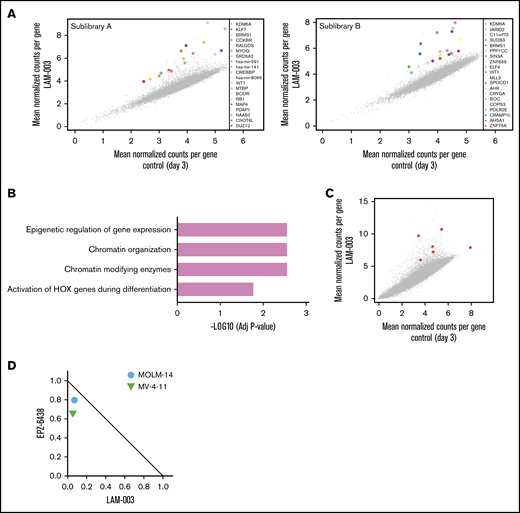
CRISPR identifies epigenetic regulation as a key determinant of LAM-003 sensitivity. (A) Scatter plot showing enrichment of the top 20 normalized sgRNA read counts in vehicle and LAM-003–treated CRISPR pools in GeCKO sublibraries A and B. (B) Gene ontology analysis of top 20 sgRNAs. (C) Scatter plot showing enrichment of normalized sgRNA read count of KDM6A in vehicle and LAM-003–treated CRISPR pools from the combined A and B GeCKO sublibraries. Six individual sgRNAs used for targeting KDM6A are shown in red. (D) Normalized isobologram at the EC75 of two FLT3-ITD–harboring cell lines treated with the EZH2 inhibitor EPZ6438 for 4 days followed by the combination of EZH2 inhibitors and LAM-003 for an additional 72 hours before viability was assayed by using CellTiter-Glo. Data points are the average of 2 independent experiments, each performed in duplicate.
CRISPR identifies epigenetic regulation as a key determinant of LAM-003 sensitivity. (A) Scatter plot showing enrichment of the top 20 normalized sgRNA read counts in vehicle and LAM-003–treated CRISPR pools in GeCKO sublibraries A and B. (B) Gene ontology analysis of top 20 sgRNAs. (C) Scatter plot showing enrichment of normalized sgRNA read count of KDM6A in vehicle and LAM-003–treated CRISPR pools from the combined A and B GeCKO sublibraries. Six individual sgRNAs used for targeting KDM6A are shown in red. (D) Normalized isobologram at the EC75 of two FLT3-ITD–harboring cell lines treated with the EZH2 inhibitor EPZ6438 for 4 days followed by the combination of EZH2 inhibitors and LAM-003 for an additional 72 hours before viability was assayed by using CellTiter-Glo. Data points are the average of 2 independent experiments, each performed in duplicate.
Discussion
The recent approval of midostaurin50 and gilteritinib51 in relapsed and refractory AML has offered renewed hope for this difficult-to-treat disease.52 Nonetheless, the acquisition of primary and secondary resistance curtails the long-term efficacy of such agents,10 and novel therapeutic agents are needed to overcome such resistance mechanisms. Here, we describe the pharmacologic activity of LAM-003, an orally bioavailable HSP90i in preclinical models of FLT3-ITD AML.
There are distinct differences in the mechanism of inhibition of oncogenic FLT3 by FLT3i compared with LAM-003. FLT3i block signaling through direct binding to the FLT3 receptor kinase domain, and thus mutations in the FLT3 TKD can confer resistance by preventing binding of FLT3i to the adenosine triphosphate–binding pocket.53 In contrast, LAM-003 disrupts the HSP90-FLT3 association that is crucial for the stability of mutated FLT3, leading to degradation and resulting in cell death.19 This notion has been shown in non-AML cell lines exogenously expressing various FLT3 TKD mutants treated with an HSP90i.19,25 To the best of our knowledge, our study is the first to show the efficacy of an HSP90i, LAM-003, in FLT3-ITD AML cell lines and primary AML cells that are refractory to FLT3i. Moreover, the finding that 22% of FLT3-ITD AML patients whose disease progressed after FLT3i was associated with the emergence of secondary mutations (D835/I836)8 highlights a patient population that has no treatment options and which may benefit from LAM-003 treatment.
In addition to mutations in the FLT3 TKD, the bone marrow provides a niche which limits the efficacy of FLT3i because extrinsic (stromal) factors provide survival signaling in AML cells that mediate resistance.10,13,14 Moreover, levels of plasma FLT3 ligand, which also confers resistance to FLT3i, are increased in patients with AML after chemotherapy,54 further limiting FLT3i efficacy. Using cell culture media that replicate these conditions,13 we confirmed that FLT3i efficacy was reduced in FLT3-ITD AML cells treated in media from stromal cells, yet LAM-003 retained efficacy under these conditions. Extrapolating these findings to the clinic, LAM-003 is expected to be effective in FLT3-ITD patients with AML who are relapsed or refractory to FLT3i due to a protective bone marrow microenvironment.
We further showed that LAM-003 exhibits synergistic activity with existing standard-of-care agents, consistent with the action of other HSP90i,55-57 and which will also be critical to combat resistance in the clinic. Importantly, we have discovered highly synergistic activity of LAM-003 with venetoclax. Based on our mechanistic studies, we attribute the synergistic activity of LAM-003 to block venetoclax induction of MCL-1, a known mechanism for venetoclax resistance.42 Although partial reduction of MCL-1 has been observed with other HSP90i,58,59 we found that the combination leads to complete degradation of MCL-1. Our findings are consistent with a model whereby the combination of LAM-003 and venetoclax leads to degradation of AKT, thereby abolishing the inhibitory phosphorylation of GSK3β(Ser9), activating GSK3β,41 and in turn targeting MCL-1 for degradation in a SCFFBW7-dependent manner.60 The finding that AKT inhibitors also synergized with venetoclax further supports the argument that LAM-003 reduces MCL-1 expression in an AKT-dependent manner. In addition, ERK phosphorylation of MCL1 leads to its stabilization,61 and the finding that LAM-003 and venetoclax result in loss of ERK activity provides a parallel mechanism for MCL-1 loss (supplemental Figure 2I). Our findings are consistent with studies showing that cotargeting of MCL-1 and BCL-2 in AML cells is synergistic.42,62,63 Moreover, because MCL-1 also confers resistance to FLT3i cytarabine and daunorubicin,64 we expect the LAM-003 and venetoclax combination to be effective at thwarting resistance, especially as LAM-003 retains potent single-agent activity in venetoclax-resistant cells in vitro and in vivo. Venetoclax exhibited modest single-agent clinical activity in relapsed/refractory patients,44 and in a follow-up study evaluating somatic mutations in AML patients treated with venetoclax, the presence of FLT3-ITD was associated with resistance.65 Our findings suggest that the combination of LAM-003 and venetoclax may improve clinical response rates in a relapsed/refractory population of FLT3-ITD AML. Because synergy of LAM-003 and venetoclax correlated with high basal BCL-2 (but not MCL-1) levels, this suggests a potential clinical biomarker to assess for the combination.
To identify unique biomarkers for LAM-003 activity, we performed a genome-wide, CRISPR-mediated, loss-of-function screen and identified many hits that have known roles in leukemogenesis and are driver mutations in AML.66 Gene ontology analysis highlighted epigenetic regulation as a key determinant of LAM-003 sensitivity, confirmed through interrogation of KDM6A and EZH2, which have opposing roles on H3K27 methylation to regulate gene transcription. LAM-003 synergy with EZH2 inhibitors may be of relevance in the AML population with low EZH2 levels (and thereby low H3K27me3 levels), a population that historically has a poor prognosis and significantly reduced overall survival compared with patients with high H3K27me3.67 In addition, our data suggest that the combination of LAM-003 and EZH2 inhibitor may be effective in patients with wild-type EZH2 status. Intriguingly, EZH2 is located on chromosome 7q,68 which was found to be deleted in 2 of 3 patients with AML (17 total evaluable patients) who obtained a complete remission with an incomplete blood count recovery when treated with the HSP90i alvespimycin.27 We will monitor the status of KDM6A and EZH2 in patients during our phase 1 trial to determine their utility as predictive biomarkers that correlate with LAM-003 response.
From a clinical perspective, LAM-003 meets prerequisite requirements for evaluation in a genetically defined population of FLT3-ITD AML patients who do not respond to therapy with FLT3i. LAM-003A, the active drug, is well tolerated as shown by extensive preclinical safety studies in mice, rats, and monkeys and in a dose escalation study in 26 patients with solid tumors, which enabled the selection of a recommended phase 2 dose.31 At this dose (480 mg/d), no safety issues were noted, and pharmacokinetic variables showed that trough levels remain above the threshold required for constitutive target suppression. Moreover, LAM-003 is orally bioavailable, results in similar exposure to LAM-003A, and is suitable for daily dosing but with a more acceptable dosage form. Based on these findings, LAM-003 is currently undergoing clinical evaluation in patients with AML to determine a recommended dose and to monitor safety, pharmacokinetic and pharmacodynamic variables, and preliminary efficacy.
Acknowledgments
The authors thank Sharyn D. Baker for providing the MOLM-13-luc and MOLM-13-RES D835Y-luc AML cells and Monica L. Guzman for insightful comments.
This work was fully supported by AI Therapeutics and in part by the DeLuca Center for Innovation in Hematology Research at Yale Cancer Center (S.H.).
Authorship
Contribution: N.B., H.L., P.R.Y., P.B., T.X., and J.R. conceived and directed the project; N.B., S.L., S.G., and J.E.G. designed and conducted the experiments; M.H. performed the bioinformatics analyses; S.L. oversaw the in vivo experiments with assistance from X.Z. and B.Z.C. for the venetoclax-resistant in vivo study; M.A., B.Z.C., and S.H. provided resources and expertise; N.B. and H.L. wrote the manuscript; and S.L., J.G., S.G., P.R.Y., P.B., and M.H. contributed to the writing of the manuscript.
Conflict-of-interest disclosure: N.B., S.L., S.G., J.E.G., P.B., M.H., P.R.Y., and H.L. are employees at AI Therapeutics. T.X. is on the AI Therapeutics advisory board. J.R. is a Director of AI Therapeutics. AI Therapeutics is the owner of LAM-003/LAM-003A patents. The remaining authors declare no competing financial interests.
Correspondence: Neil Beeharry, AI Therapeutics, 530 Whitfield St, Guilford, CT 06437; e-mail: nbeeharry@ai-thera.com.

