

This article has a companion
Pro (but with a “little but”)
Over the past 10 years, next-generations sequencing (NGS) has allowed major advances in unraveling the complex and diverse molecular signature of myelodysplastic syndromes (MDS).1-4 As a result, over 50 recurrent mutations have been identified in MDS.1,2 In the majority of cases, at least 1 mutation (frequently 2-4) can be identified with targeted sequencing in an individual MDS patient.1,2 There is not one single mutation that is 100% pathognomonic for MDS, and we have learned that mutations in MDS cluster in functional genetic groups. While NGS has undoubtedly played a relevant role in the identification of novel mutations in MDS, it is legitimate to ask whether and how NGS can be beneficial for an individual MDS patient. Should we perform NGS for our MDS patients in routine clinical practice, and if so, when and at which technical level? Our answer is yes (in the vast majority of patients) and includes some caveats that will be discussed.
Diagnosing MDS
Conventional diagnosis of MDS is based on cytology, histology, and cytogenetics. So far, NGS cannot replace this diagnostic workup and should not serve as a “stand-alone” diagnostic tool. However, the mutational status of SF3B1 is now included as a parameter of the revised World Health Organization (WHO) classification of myeloid neoplasms and is therefore recommended in all low-risk cases given the excellent prognosis.5 In case a SF3B1 mutation is present, only 5% of ring sideroblasts (RS) instead of 15% need to be detected in order to diagnose MDS with RS.5 Appropriate conventional morphological and cytogenetic workup is mandatory for patients with cytopenia of unknown origin. Nevertheless, NGS can add helpful diagnostic information (Figure 1). For example, if a bone marrow aspirate or biopsy specimen reveals mild cytopenia and dysplasia, it can be challenging even for experienced morphologists to distinguish reactive changes from true MDS.6 Here, NGS including a large panel of genes can help to proof or exclude a clonal disease including clonal cytopenia of undetermined significance, which can have a significant impact on subsequent surveillance strategies.7 Additionally, clonal hematopoiesis of indeterminate potential (CHIP), which describes a novel entity in which somatic mutations associated with myeloid malignancies are found in blood or bone marrow cells of people, where other criteria for hematologic neoplasia (eg, cytopenia or dysplasia) are not met, needs to be clearly distinguished from MDS.8-10 Besides MDS defining criteria (eg, cytopenia or dysplasia), a greater number of mutations with a higher variant allelic frequency (VAF) are more molecularly supportive for the diagnosis of MDS than CHIP.11 The discrimination of hypoplastic MDS from aplastic anemia (AA) can be difficult (eg, mutations in PIGA are relatively frequent in AA but very rare in MDS).12,13 Here, a greater number of mutations with a higher VAF are rather supportive for the diagnosis of MDS. CEBPa, NPM1, and FLT3 mutations are acute myeloid leukemia (AML)–defining mutations and might be important to look for in cases with excess of blasts, especially when close to the 20% threshold, where intensive chemotherapy is an option. Identification of a KIT mutation can be helpful in suspected cases with systemic mastocytosis with associated hematological disease (SM-AHD) (Figure 2). Similarly, STAT3 mutational screening can be useful for the detection of large granular lymphocyte leukemia associated cytopenia.14 It is likely that more molecular markers will find their way into the WHO classification of MDS in the future.
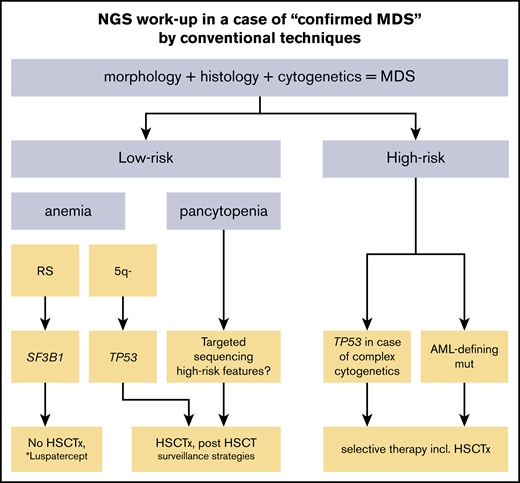
Schematic overview of the NGS workup and potential clinical consequences in case of confirmed MDS by conventional techniques. AML-defining mutations include CEBPA, NPM1, and FLT3-ITD. HSCTx, allogeneic hematopoietic stem cell transplantation (possibly with preceding chemotherapy); SF3B1, mutations in SF3B1; TP53, mutations in TP53. *Currently not approved.
Schematic overview of the NGS workup and potential clinical consequences in case of confirmed MDS by conventional techniques. AML-defining mutations include CEBPA, NPM1, and FLT3-ITD. HSCTx, allogeneic hematopoietic stem cell transplantation (possibly with preceding chemotherapy); SF3B1, mutations in SF3B1; TP53, mutations in TP53. *Currently not approved.
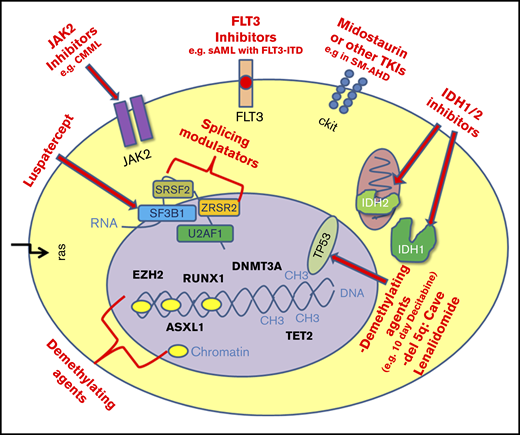
Schematic overview of mutations in MDS as potential therapeutic targets. CMML, chronic myelomonocytic leukemia; sAML, secondary acute myeloid leukemia; TKIs, tyrosine kinase inhibitors.
Schematic overview of mutations in MDS as potential therapeutic targets. CMML, chronic myelomonocytic leukemia; sAML, secondary acute myeloid leukemia; TKIs, tyrosine kinase inhibitors.
Prognostication of MDS
While the International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS-R) are the gold standard for prognostication in MDS,15,16 the presence of some molecular markers incl. SF3B1 (as noted above) or TP531 are known to have an independent prognostic effect. In fact, the presence of TP53 adds to the prognosis of patients with deletion of chromosome 5q (del5q) in low-risk MDS and complex karyotype in high-risk MDS.17,18 Therefore, we recommend routine testing of the latter in patients with low-risk MDS (SF3B1 all patients and TP53 in del5q only) or high-risk disease (TP53 in complex karyotype). Routine testing of other poor-risk mutations like RUNX1 or ASXL1 might help to further identify patients with a worse prognosis of all stages. We perform testing regularly in lower-risk cases where an intensification of disease surveillance followed by putative allogeneic hematopoietic stem cell transplantation (HSCT) might be an option. NGS techniques can analyze a large number of genes, almost at the same cost irrespective of the gene number. Therefore, this is often the preferred sequencing method, as it gives additional information about the number of mutated genes, which has been shown to be prognostic (ie, the more mutated genes a patient has, the worse the outcome).1,2 Ongoing efforts by an international MDS consortium try to integrate molecular data within the IPSS and IPSS-R. Furthermore, NGS has been used to predict the AML risk in healthy individuals with clonal hematopoiesis.19
Prediction of response and outcome to conventional therapy
Currently, treatment options in MDS are limited and include growth factors, lenalidomide, hypomethylating agents (HMAs), and HSCT. The number of mutations has been linked to response to erythropoiesis-stimulating agent (ESA) in low-risk MDS.20 Luspatercept is a novel agent used to treat anemia in low-risk MDS patients failing ESA treatment.21 The drug directly targets the transforming growth factor β pathway. In a phase 2 trial, SF3B1-mutated patients were more likely to respond to luspatercept.21 While lenalidomide is standard for patients with isolated del5q, there is evidence that patients with del5q who also harbor a TP53 mutation (up to 20%) are less likely to respond to lenalidomide and have a worse prognosis.22
Interestingly, retrospective data in higher-risk MDS and AML patients treated with dose-intense decitabine (20 mg/m2 over 10 days) suggest that patients with TP53 mutations were more likely to respond to this intensive epigenetic treatment than patients with other mutations.23 Furthermore, TET2 mutational status has been identified to be predictive for response to standard HMAs, although the data are conflicting.24-26 This is also because the higher response rate did not translate into a survival benefit for TET2-mutated patients,24,25 and again, prospective data are missing. Therefore, we would not recommend routine testing before treatment with HMAs only. Last, but not least, the outcome of HSCT can be impacted by the molecular profile of a patient. While retrospective data of smaller cohorts have led to conflicting results, data from larger cohorts revealed TP53 and RAS mutations as independent adverse markers in HSCT.27-29 Patients with a high-risk mutational profile might benefit the most from intensified posttransplant surveillance strategies.30
Novel targeted therapy
Precision oncology, defined as molecular profiling of tumors to identify targetable alterations, is very appealing, as it combines the hopes for a more effective treatment with less toxicity compared with conventional treatment. Due to the molecular heterogeneity of MDS, a potential target needs to be identified individually in each MDS patient. In MDS, targeted therapies are just beginning to emerge (eg, IDH1/2 inhibitors).31 In a phase 1/2 trial, enasidenib (AG221), a selective IDH2 mutant inhibitor, was used as monotherapy in refractory/relapsed AML patients, leading to an overall response rate of 38%.32 In MDS, ∼4% to 5% of patients carry an IDH2 mutation, and 2% to 3% carry an IDH1 mutation.1,2,33 IDH1 and IDH2 inhibitors are currently not approved for AML or MDS patients in Europe, but enasidenib has received approval by the US Food and Drug Administration for elderly relapsed/refractory AML patients. It is likely that the response data can be transferred to patients with MDS, especially when progressing to sAML. However, several trials for IDH1- and IDH2-mutant AML and MDS patients are currently ongoing, making molecular screening with NGS necessary for patient enrollment. Other examples of potential targeted therapies are FLT3-ITD inhibitors in sAML or so-called splicing modulators in lower-risk MDS. Splicing gene mutations are among the most frequent mutations found in MDS (eg, SF3B1, SRSF2, U2AF1, and ZRSR2 mutations).1,2,34 Splicing modulators are intended to induce “splicesomal sickness” and consequently cell death. Currently, treatment with splicing modulators is only possible within clinical trials, and it will be interesting to see the outcome of these trials. Other ongoing “targeted approaches” involve JAK-2 inhibitors in MDS/myeloproliferative neoplasm cases and as well as KIT modulation in SM-AHD. It will be interesting to see whether mutated TP53 proves to be a promising target, as early results with Prima-1Met (APR-246), an activator of TP53 function, suggest.35 In general, clinical trials with targeted therapies are currently still rare and only possible with the availability of NGS.
Further aspects
Over the past decade, we have gained novel insights into the genetic origin of inherited myeloid malignancies.36,37 Mutations in GATA2, RUNX1, DDX41, and ETV6 are examples of germline mutations associated with MDS and AML.36,37 The detection of germline mutations has a significant impact not only for treatment decisions but also for the patient’s family. Obtaining a detailed family history, testing germline material (eg, skin fibroblasts), and genetic counseling of patients and families should go hand in hand. Last, but not least, NGS has evolved as a very attractive tool for measurable residual disease (MRD) monitoring after chemotherapy or HSCT.38-40 We acknowledge that access to NGS is limited in some countries and is also associated with a clear economic burden. Thus, a targeted sequencing approach that accounts for the diagnostic/clinical scenario can be sensible, keeping the potential therapeutic implications in mind (Figures 1 and 2). Besides reporting the type and location of the mutation, the VAF should also be included in NGS report. Is NGS necessary for every MDS patient? We believe that NGS should be performed in the vast majority of MDS patients also because we as clinicians have to learn the impact and relevance of this clinical information on a patient-by-patient basis. However, NGS testing outside clinical trial options is not helpful for patients who have a very short life expectancy related to another underlying critical disease and/or very advanced disease.
In summary, we would advocate for NGS (commonly with targeted approach) in the vast majority of MDS patients, as NGS results can be helpful with the diagnosis, prognostication, and treatment response predication in MDS patients (Figures 1 and 2). In addition, NGS opens the opportunity of targeted therapies (in clinical trials). The relevance of NGS is likely to increase in the next few years with further implications such as MRD monitoring. With NGS becoming cheaper over the years, our “little but” will become even smaller.
Acknowledgments
U.P. received support from Deutsche Jose-Carreras Leukämiestiftung and Boll-Stiftung.
Authorship
Contribution: U.P. and F.T. both performed a literature search and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Uwe Platzbecker, Medical Clinic and Policlinic 1, Hematology and Cellular Therapy, Leipzig University Hospital, Leipzig, Johannisallee 32 A, 04103 Leipzig, Germany; e-mail: uwe.platzbecker@medizin.uni-leipzig.de.

