

Key Points
Cas9 editing of the γ-globin gene promoters in hematopoietic stem cells (HSCs) increases red cell HbF by ≤40%.
No deleterious effects on hematopoiesis or off-target mutations were detected 16 weeks after xenotransplantation of edited HSCs.

Abstract
Induction of fetal hemoglobin (HbF) via clustered regularly interspaced short palindromic repeats/Cas9–mediated disruption of DNA regulatory elements that repress γ-globin gene (HBG1 and HBG2) expression is a promising therapeutic strategy for sickle cell disease (SCD) and β-thalassemia, although the optimal technical approaches and limiting toxicities are not yet fully defined. We disrupted an HBG1/HBG2 gene promoter motif that is bound by the transcriptional repressor BCL11A. Electroporation of Cas9 single guide RNA ribonucleoprotein complex into normal and SCD donor CD34+ hematopoietic stem and progenitor cells resulted in high frequencies of on-target mutations and the induction of HbF to potentially therapeutic levels in erythroid progeny generated in vitro and in vivo after transplantation of hematopoietic stem and progenitor cells into nonobese diabetic/severe combined immunodeficiency/Il2rγ−/−/KitW41/W41 immunodeficient mice. On-target editing did not impair CD34+ cell regeneration or differentiation into erythroid, T, B, or myeloid cell lineages at 16 to 17 weeks after xenotransplantation. No off-target mutations were detected by targeted sequencing of candidate sites identified by circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq), an in vitro genome-scale method for detecting Cas9 activity. Engineered Cas9 containing 3 nuclear localization sequences edited human hematopoietic stem and progenitor cells more efficiently and consistently than conventional Cas9 with 2 nuclear localization sequences. Our studies provide novel and essential preclinical evidence supporting the safety, feasibility, and efficacy of a mechanism-based approach to induce HbF for treating hemoglobinopathies.
Introduction
Sickle cell disease (SCD) and β-thalassemia are common disorders caused by HBB gene mutations that alter quantity or quality of the β-globin subunit of adult hemoglobin (HbA, α2β2).1,2 Severely affected individuals experience multiorgan damage, with substantial morbidity and early mortality. Αllogeneic hematopoietic stem cell (HSCs) transplantation can be curative but carries high risk of severe toxicities, particularly for patients who lack fully histocompatible donors.3 Hence, new methods for autologous gene therapy are being sought.
Genome editing of patient HSCs by clustered regularly interspaced short palindromic repeats (CRISPR)–Cas9 nucleases represents a promising approach for genetic correction of β-hemoglobinopathies.4-6 These nucleases introduce targeted DNA double-stranded breaks (DSBs) that can be exploited therapeutically through 2 general cellular DNA damage repair strategies. First, HBB mutations can be corrected via homology-directed repair (HDR).5,7-12 Second, fetal hemoglobin (HbF, α2γ2) can be induced in adult red blood cells (RBCs) by using nonhomologous end-joining (NHEJ) mediated mutations to disrupt noncoding DNA regulatory elements that repress transcription of the genes encoding γ-globin (HBG1 and HBG2) postnatally.4,13-19 Numerous clinical studies show that elevated HbF production is associated with reduced morbidity and mortality in SCD and β-thalassemia.20,21 In extreme cases, a rare, benign genetic condition called hereditary persistence of HbF (HPFH) induces high-level pancellular HbF expression in RBCs and eliminates the pathologies of coinherited β-hemoglobinopathies.22 Therapeutically, the induction of HbF by NHEJ may offer several advantages over direct HBB repair for treating β-hemoglobinopathies. First, NHEJ is the dominant DNA DSB repair pathway and is active in all phases of the cell cycle, which is particularly relevant to editing quiescent HSCs. Second, correction of the SCD mutation via HDR is accompanied by undesired NHEJ-mediated insertion/deletion (indel) mutations in cis or trans.7,23 Third, in contrast to HDR, NHEJ does not require delivery of an exogenous DNA donor template, which introduces additional technical challenges in manufacturing. Lastly, genome editing to induce HbF provides a single generally applicable therapeutic strategy for many different β-hemoglobinopathy mutations, whereas correction by HDR would require mutation-specific optimization.
Several HPFH mutations occur in a region 118 to 114 nt upstream of the HBG1 and HBG2 transcription start sites and disrupt a cognate-binding element for the γ-globin gene repressor BCL11A (TGACC).24,25 Previously, we targeted this region in CD34+ hematopoietic stem and progenitor cells (HSPCs) by lentiviral expression of Cas9 and associated single guide RNAs (sgRNAs) followed by in vitro differentiation.16 The percentage of HbF (%HbF) was increased to potentially therapeutic levels in the RBC progeny of most CD34+ cells with on-target edits. Here we advance that proof-of-concept study by achieving several essential requirements for clinical translation, including transient Cas9:sgRNA delivery to HSPCs, high-level editing in human HSCs capable of multilineage engraftment after transplantation into immunodeficient mice, and absence of detectable off-target mutations or deleterious hematopoietic effects. Therefore, Cas9 ribonucleoprotein (RNP)–mediated disruption of the BCL11A repressor binding site in the promoters of HBG1 and HBG2 is a potentially feasible and safe therapeutic strategy for treating SCD and β-thalassemia.
Methods
Human subjects research
Plerixafor-mobilized CD34+ cells from patients with SCD were collected according to the protocol Peripheral Blood Stem Cell Collection for Sickle Cell Disease Patients (www.clinicaltrials.gov identifier #NCT03226691), which was approved by the human subject research institutional review boards at the National Institutes of Health and St. Jude Children’s Research Hospital. All patients provided informed consent.
Animal care
Mice were housed and handled in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal experiments were carried out in accordance with a protocol (Genetic Tools for the Study of Hematopoiesis) approved by the institutional animal care and use committee of the St. Jude Children’s Research Hospital or Boston Children’s Hospital.
Cell culture, editing, and xenotransplantation
The antibodies used in this study are listed in supplemental Table 1. The cytokines used are listed in supplemental Table 2. The oligonucleotides used are listed in supplemental Table 3. The isolation, editing, and analysis of CD34+ cells before and after xenotransplantation are described in the supplemental Methods.
Results
Optimization of HBG1/HBG2 promoter editing in human CD34+ cells via Cas9:sgRNA RNPs
We edited human CD34+ HSPCs by electroporating RNP complexes of Cas9:sgRNA-1 targeting the BCL11A consensus motif at position −118 to −114 in the HBG1/HBG2 gene promoters (Figure 1A). Initial studies were performed using the Neon Transfection System (see supplemental Methods). We titrated Cas9 and sgRNA-1 with or without 2′-O-methyl modifications and 3′-phosphorothioate internucleotide linkages shown previously to improve gene editing rates in primary cells26 and electroporated the RNP complexes into adult human peripheral blood–mobilized CD34+ HSPCs. Transfected cells were cultured for 4 days and then analyzed for small indels by next-generation sequencing (NGS) of a 318-bp polymerase chain reaction (PCR) fragment centered around the RNP cleavage site (Figure 1B; supplemental Figure 1A-B). The maximum indel frequencies observed were ∼45% with unmodified sgRNA and 80% with chemically modified sgRNA. After electroporation, cell viability declined with increasing RNP concentration but remained >80% under all conditions (supplemental Figure 1C-E). Importantly, the 9 most frequently observed indel alleles, and presumably most others, disrupt the BCL11A-binding site and therefore are predicted to derepress the associated γ-globin genes (Figure 1C).
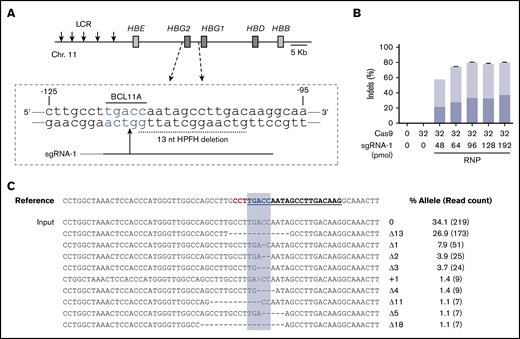
Cas9:sgRNA RNP disruption of a transcriptional repressor binding motif in the γ-globin gene promoters. (A) The extended β-globin locus, showing the target BCL11A binding motif in the promoters of the genes encoding γ-globin (HBG2 and HBG1). The 13-nt HPFH deletion is indicated by a dashed black line. sgRNA-1 target is represented by a black line, with a vertical arrow indicating the predicted site of Cas9 dsDNA cleavage and the protospacer adjacent motif (PAM) sequence indicated in gray. (B) Optimization of the Cas9:sgRNA-1 ratio on indel formation, using the Neon Transfection System. RNPs were generated by incubating the indicated amounts of Cas9 and sgRNA in 5 μL of 10 mM HEPES, 150 mM NaCl for 30 min at room temperature. Here and in subsequent experiments, sgRNA-1 was chemically modified to enhance stability. The RNPs were mixed with 2 × 105 CD34+ cells in T buffer in a final volume of 10 μL, electroporated using a Neon Transfection System at 1600 V, with 3 pulses of 10 ms, cultured for 4 days, and then analyzed for indel formation by high-throughput sequencing of PCR products generated using primers located ∼150 bp on either side of the RNP cleavage site. The graphs show the mean ± standard deviation (SD) indel frequency on the y-axis (n = 3 biological replicates). Dark blue indicates the 13-nt human HPFH mutation; light blue represents all other indels. (C) Sequence alignment of the most common mutant alleles. The sgRNA-1 sequence is underlined and bolded, the PAM sequence is in red, and the BCL11A-binding motif is highlighted in blue. Deletions are represented by dashes. The percentage of each mutation observed is shown on the right, with the NGS read counts in parentheses. LCR, locus control region.
Cas9:sgRNA RNP disruption of a transcriptional repressor binding motif in the γ-globin gene promoters. (A) The extended β-globin locus, showing the target BCL11A binding motif in the promoters of the genes encoding γ-globin (HBG2 and HBG1). The 13-nt HPFH deletion is indicated by a dashed black line. sgRNA-1 target is represented by a black line, with a vertical arrow indicating the predicted site of Cas9 dsDNA cleavage and the protospacer adjacent motif (PAM) sequence indicated in gray. (B) Optimization of the Cas9:sgRNA-1 ratio on indel formation, using the Neon Transfection System. RNPs were generated by incubating the indicated amounts of Cas9 and sgRNA in 5 μL of 10 mM HEPES, 150 mM NaCl for 30 min at room temperature. Here and in subsequent experiments, sgRNA-1 was chemically modified to enhance stability. The RNPs were mixed with 2 × 105 CD34+ cells in T buffer in a final volume of 10 μL, electroporated using a Neon Transfection System at 1600 V, with 3 pulses of 10 ms, cultured for 4 days, and then analyzed for indel formation by high-throughput sequencing of PCR products generated using primers located ∼150 bp on either side of the RNP cleavage site. The graphs show the mean ± standard deviation (SD) indel frequency on the y-axis (n = 3 biological replicates). Dark blue indicates the 13-nt human HPFH mutation; light blue represents all other indels. (C) Sequence alignment of the most common mutant alleles. The sgRNA-1 sequence is underlined and bolded, the PAM sequence is in red, and the BCL11A-binding motif is highlighted in blue. Deletions are represented by dashes. The percentage of each mutation observed is shown on the right, with the NGS read counts in parentheses. LCR, locus control region.
Gene editing induces HbF expression in cultured erythroid cells
We cultured gene-edited CD34+ HSPCs from 2 donors under erythroid differentiation conditions and measured the HbF expression in late-stage erythroblasts. The mean indel frequency measured was 63% ± 18% (Figure 2A). As usual for in vitro–derived erythroblasts, background HbF expression was high; ∼50% of the cells stained with HbF antibody (termed F-cells), and HbF protein constituted ~10% of the total Hb in cell lysates (Figure 2B-C; supplemental Figure 2). Nonetheless, gene editing of the HBG1 and HBG2 gene promoters raised %F-cells and %HbF protein significantly. Editing did not alter the expression of erythroid maturation markers Band3 or CD49d (Figure 2D) or the conversion of nucleated erythroblasts to anucleate reticulocytes (Figure 2E).
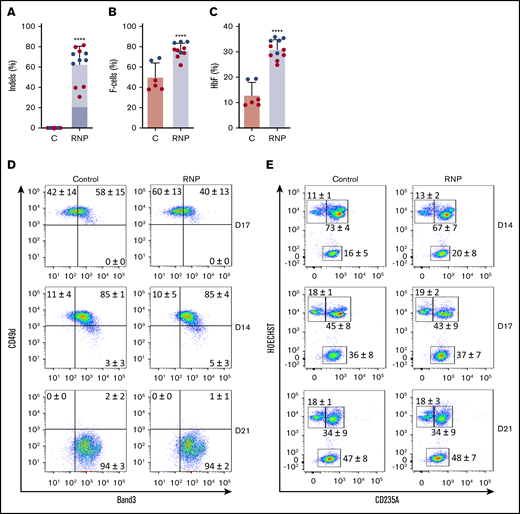
Gene editing induces HbF expression in cultured erythroid cells. G-CSF–mobilized CD34+ cells were edited with Cas9:sgRNA-1 RNP using the Neon Transfection System (supplemental Methods). They were then grown in culture for 21 days under conditions that support erythroid differentiation. Data show studies from 2 different CD34+ cell donors (blue or red), with each dot representing separate experiments. (A) Indel frequencies in gene-edited (RNP) and control (C) groups determined 4 days after editing. Dark blue indicates the 13-nt human HPFH mutation; light blue represents all other indels. (B) Cells immunostaining for hemoglobin F (F-cells) measured by flow cytometry at culture day 21. (C) The %HbF in day 21 erythroid cell lysates determined by ion-exchange high-performance liquid chromatography (HPLC). The bar charts in panels A-C show the results as the mean ± SD. **** P < .0001 (unpaired Student t test). (D) Erythroid maturation kinetics at culture days 7, 14, and 21, as determined by immunoflow cytometry measurement of CD49d and Band3 expression on CD235a+ erythroid cells. (E) Enucleated cell fractions at culture days 14, 17, and 21, as determined by flow cytometry for CD235a and the cell-permeable DNA dye Hoechst 333412. The numbers in each quadrant show the mean percentages ± SDs. The data reflect studies of CD34+ cells from 2 different donors in 7 independent experiments.
Gene editing induces HbF expression in cultured erythroid cells. G-CSF–mobilized CD34+ cells were edited with Cas9:sgRNA-1 RNP using the Neon Transfection System (supplemental Methods). They were then grown in culture for 21 days under conditions that support erythroid differentiation. Data show studies from 2 different CD34+ cell donors (blue or red), with each dot representing separate experiments. (A) Indel frequencies in gene-edited (RNP) and control (C) groups determined 4 days after editing. Dark blue indicates the 13-nt human HPFH mutation; light blue represents all other indels. (B) Cells immunostaining for hemoglobin F (F-cells) measured by flow cytometry at culture day 21. (C) The %HbF in day 21 erythroid cell lysates determined by ion-exchange high-performance liquid chromatography (HPLC). The bar charts in panels A-C show the results as the mean ± SD. **** P < .0001 (unpaired Student t test). (D) Erythroid maturation kinetics at culture days 7, 14, and 21, as determined by immunoflow cytometry measurement of CD49d and Band3 expression on CD235a+ erythroid cells. (E) Enucleated cell fractions at culture days 14, 17, and 21, as determined by flow cytometry for CD235a and the cell-permeable DNA dye Hoechst 333412. The numbers in each quadrant show the mean percentages ± SDs. The data reflect studies of CD34+ cells from 2 different donors in 7 independent experiments.
Gene editing of HSCs induces HbF expression in erythroid progeny generated in vivo
To assess our capacity to edit repopulating HSCs, we electroporated CD34+ cells with Cas9:sgRNA-1 RNP and transplanted them into nonobese diabetic/severe combined immunodeficiency/Il2rγ−/−/KitW41/W41 (NBSGW) mice, an immunodeficient strain that supports the development of human erythroid precursors without conditioning.27 We measured the fraction of engrafted human cells and their indel frequencies serially in blood and in bone marrow after the mice were euthanized at 17 weeks, when donor CD34+ cells present derive mainly from HSCs.27 No significant differences in chimerism occurred between edited and nonedited donor-derived CD45+ hematopoietic cell populations (Figure 3A). Chimerism levels were similar for edited and nonedited donor T cells (hCD45+/CD3+), B cells (hCD45+/CD19+), myeloid cells (hCD45+/CD33+), and erythroid cells (CD45−/CD235a+) in bone marrow at 17 weeks (Figure 3B).
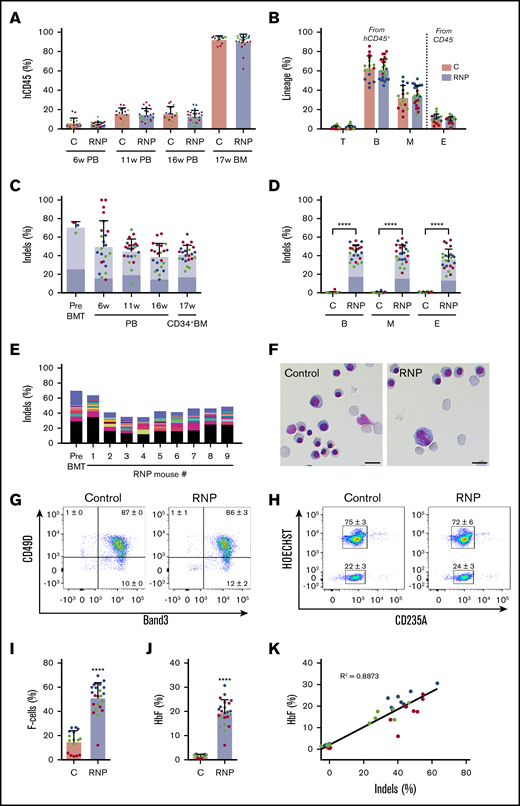
Xenotransplantation of normal gene-edited CD34+cells into NBSGW mice. Peripheral blood (PB) CD34+ cells were edited with Cas9:sgRNA-1 RNP using the Neon Transfection System and then transplanted into NBSGW mice via tail-vein injection. Donor cell controls (C) were processed in parallel, but not electroporated. Donor-cell progeny were analyzed in recipient peripheral blood at 6, 11, and 16 weeks and in bone marrow (BM) at 17 weeks after transplantation. Data are from 3 independent experiments with CD34+ cells from different donors (red, blue, or green). Each dot in graphs represents a separate mouse. (A) Normalized human chimerism in peripheral blood and bone marrow, shown as the percentage of human (h) CD45+ cells. (B) Human T (CD3+), B (CD19+), and myeloid (M) (CD33+) cells shown as percentages of the human CD45+ population in bone marrow at 16 weeks. Erythroid chimerism (E) is shown as the percentage of human CD235a+ cells within the CD45− population (mouse and human). (C) The indel fraction 4 days after editing (Pre BMT) and as measured serially after xenotransplantation. Dark blue represents the 13-nt human HPFH mutation; light blue represents all other indels. (D) Indels in specific hematopoietic lineages. Dark blue represents the 13-nt human HPFH mutation; light blue represents all other indels. (E) The most frequent indels in donor 1 CD34+ cells before bone marrow transplantation and in the 16-week bone marrow of mouse recipients. Unique indels are shown with a different color, with the bar size representing the relative abundance. The 13-nt HPFH mutation is represented in black. All indels present in proportions <1% are represented collectively in blue at the top of each bar. (F) Morphology of human CD235a+ erythroblasts in bone marrow of recipient mice transplanted with RNP-edited or control CD34+ cells. The images were acquired with a Nikon Eclipse Ni microscope with a 60× objective and Nikon NIS-Elements software (scale bars, 10 μm). (G) Flow cytometry for the erythroid maturation markers CD49d and Band3 on human CD235a+ erythroblasts in recipient mouse bone marrow. (H) Enucleated (HOECHST−) and nucleated (HOECHST+) human CD235a+ erythroblasts in recipient bone marrow. The numbers in panels G-H represent the mean ± SD of the percentage for the same samples analyzed in panels A-D. (I) The fraction of CD235a+ F-cells in bone marrow. (J) The %HbF in human CD235a+ erythroblasts isolated from recipient bone marrow. (K) The %HbF as a function of the indel percentage from the values plotted in panels C and J. The bar charts in panels A-D and I-J show the mean ± SD percentages for 3 biological replicates performed using CD34+ cells from 3 donors. Each dot represents a single recipient mouse, and each color represents a separate replicate study. ****P < .0001 (by unpaired Student t test).
Xenotransplantation of normal gene-edited CD34+cells into NBSGW mice. Peripheral blood (PB) CD34+ cells were edited with Cas9:sgRNA-1 RNP using the Neon Transfection System and then transplanted into NBSGW mice via tail-vein injection. Donor cell controls (C) were processed in parallel, but not electroporated. Donor-cell progeny were analyzed in recipient peripheral blood at 6, 11, and 16 weeks and in bone marrow (BM) at 17 weeks after transplantation. Data are from 3 independent experiments with CD34+ cells from different donors (red, blue, or green). Each dot in graphs represents a separate mouse. (A) Normalized human chimerism in peripheral blood and bone marrow, shown as the percentage of human (h) CD45+ cells. (B) Human T (CD3+), B (CD19+), and myeloid (M) (CD33+) cells shown as percentages of the human CD45+ population in bone marrow at 16 weeks. Erythroid chimerism (E) is shown as the percentage of human CD235a+ cells within the CD45− population (mouse and human). (C) The indel fraction 4 days after editing (Pre BMT) and as measured serially after xenotransplantation. Dark blue represents the 13-nt human HPFH mutation; light blue represents all other indels. (D) Indels in specific hematopoietic lineages. Dark blue represents the 13-nt human HPFH mutation; light blue represents all other indels. (E) The most frequent indels in donor 1 CD34+ cells before bone marrow transplantation and in the 16-week bone marrow of mouse recipients. Unique indels are shown with a different color, with the bar size representing the relative abundance. The 13-nt HPFH mutation is represented in black. All indels present in proportions <1% are represented collectively in blue at the top of each bar. (F) Morphology of human CD235a+ erythroblasts in bone marrow of recipient mice transplanted with RNP-edited or control CD34+ cells. The images were acquired with a Nikon Eclipse Ni microscope with a 60× objective and Nikon NIS-Elements software (scale bars, 10 μm). (G) Flow cytometry for the erythroid maturation markers CD49d and Band3 on human CD235a+ erythroblasts in recipient mouse bone marrow. (H) Enucleated (HOECHST−) and nucleated (HOECHST+) human CD235a+ erythroblasts in recipient bone marrow. The numbers in panels G-H represent the mean ± SD of the percentage for the same samples analyzed in panels A-D. (I) The fraction of CD235a+ F-cells in bone marrow. (J) The %HbF in human CD235a+ erythroblasts isolated from recipient bone marrow. (K) The %HbF as a function of the indel percentage from the values plotted in panels C and J. The bar charts in panels A-D and I-J show the mean ± SD percentages for 3 biological replicates performed using CD34+ cells from 3 donors. Each dot represents a single recipient mouse, and each color represents a separate replicate study. ****P < .0001 (by unpaired Student t test).
The indel frequencies of edited human donor cells declined from 70.9% ± 5.8% a few days after editing to 41.8% ± 9.4% at 17 weeks after xenotransplantation (Figure 3C), similar to what has been reported previously.14,23,28 Notably, the 13-nt HPFH mutation occurred at similar frequencies in CD34+ HSPCs before and after transplantation (Figure 3C; supplemental Figure 3). Indel frequencies were also similar in CD34+ donor cell–derived myeloid, erythroid, and B cells (Figure 3D), indicating that editing did not alter the development of these lineages from HSCs. The indel spectrum in edited cells was similar before and after transplantation, with no clonal dominance in the latter populations (Figure 3E; supplemental Figure 3).
Circulating human RBCs are short lived and difficult to detect in mouse xenotransplantation studies. Therefore, we studied immune-selected CD235a+ donor-derived erythroblasts from the bone marrow of recipient mice at 17 weeks after transplantation. We obtained mainly late-stage hemoglobinized erythroblasts and reticulocytes, with no obvious differences in morphology, expression of maturation markers, or enucleation fraction between the edited and control populations (Figure 3F-H). Notably, background %HbF was lower in control (nonedited) erythroblasts generated in vivo compared with in vitro cultures (compare Figure 2B-C to Figure 3I-J). The percentages of both HbF-immunostaining cells (F cells) and HbF protein compared with overall Hb were significantly increased in edited vs control erythroid cells generated in vivo (Figure 3I-J). Considering that the F-cell fraction was 51.3% ± 12.5% in erythroblasts with ∼42% indels (Figure 3D), it is likely that HbF was induced in all edited cells, thereby favoring therapeutic efficacy.29 The %HbF in erythroid cell lysates correlated with indel frequency (Figure 3K). These xenotransplantation studies demonstrate that it is feasible to induce erythroid HbF expression by Cas9 RNP-mediated disruption of the HBG1/HBG2 BCL11A-binding motif, with no obvious alterations in hematopoietic development in vivo.
A 4.9-kb deletion arising from simultaneous double-stranded DNA (dsDNA) cleavage at HBG1 and HBG2 on-target sites
The tandem HBG1 and HBG2 genes harbor nearly identical nucleotide sequences, including recognition sites for sgRNA-1. Simultaneous RNP-induced DSBs at both genes can result in the deletion of the intervening 4.9-kb region,16,30 leaving a single hybrid gene with HBG2 promoter sequences fused to the downstream HBG1 gene (Figure 4A). We developed 2 assays as proxies for the 4.9-kb deletion. First, we used a TaqMan quantitative PCR assay to quantify loss of DNA in the HBG2-HBG1 intergenic region ≥366 bp upstream of the RNP cleavage site (“Δ366+”). Second, we PCR-amplified exon 3 of HBG1 and HBG2, using a common primer pair, and performed NGS to quantify loss of HBG2 (“ΔHBG2”) based on a single-nucleotide difference between HBG2 and HBG1 (G vs C at cDNA position 410) (Figure 4A). Both assays showed that the 4.9-kb deletion rates were ∼30% before transplantation and 11% at 17 weeks posttransplantation (Figure 4B). By comparison, small indel rates were ∼72% and 43% before and after transplantation, respectively.
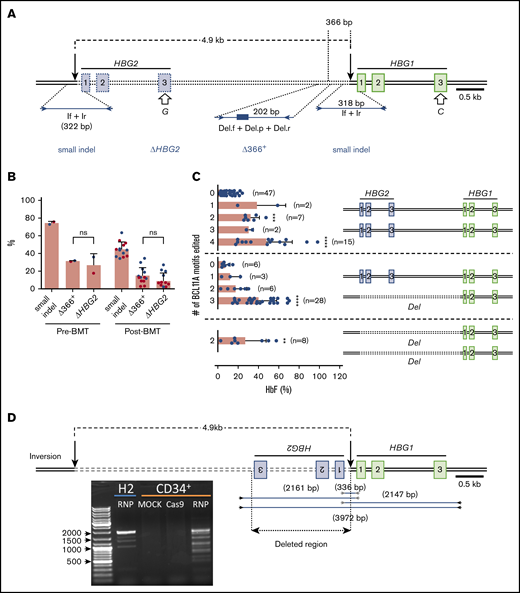
Consequences of on-target editing of HBG1 and HBG2. (A) Scale diagram of the HBG1 and HBG2 genes with exons shown as rectangles and on-target Cas9:sgRNA-1 DNA cleavage sites indicated by vertical black arrows. The genes differ by a single nucleotide in exon 3 (open arrows). Primer pair If+Ir amplifies a region surrounding the RNP cleavage site of both genes; NGS of this PCR product identified small on-target indels. Larger deletions presumed to arise from simultaneous dsDNA breaks in HBG2 and HBG1, with the loss of the intervening 4.9 kb (dotted line), were indicated by 2 proxy methods. “ΔHBG2” denotes dropout of HBG2 exon 3 based on the single-nucleotide difference between HBG2 and HBG1, determined by high-throughput sequencing of PCR products generated by a single primer pair (not shown). “Δ366+” denotes a quantitative TaqMan PCR assay that detects deletions ≥366 nt upstream of the Cas9:sgRNA-1 cleavage site in HBG1. (B) Large deletions detected by the ΔHBG2 and Δ366+ assays in CD34+ cells 4 days after editing (Pre-BMT) and 17 weeks after xenotransplantation (Post-BMT). Data reflect studies from 2 different CD34+ cell donors (blue or red dots), with each dot after bone marrow transplantation representing a single mouse. (C) Mutational analysis and %HbF in single BFU-E colonies. Each dot represents a BFU-E colony grouped by number of 4.9-kb deletions (Del) using the ΔHBG2 assay as proxy and the number of indels in the remaining BCL11A binding motifs of HBG2, HBG1, or the modified HBG1 fusion gene formed by deletion repair. Thus, heterozygosity for the 4.9-kb deletion leaves 3 potential remaining BCL11A-binding motifs and associated HBG coding regions, whereas homozygosity leaves only 2 such motifs. The bars represent the percentages of HbF as the means ± SDs. **P < .01, ***P < .001, ****P < .0001 (by unpaired Student t test). (D) Inversions resulting from simultaneous editing of HBG2 and HBG1 target sites were characterized by PCR analysis using the indicated primer pairs. The agarose gel image shows PCR products from the bulk-edited HUDEP-2 (H2) erythroid cell line and CD34+ cells. ns, not significant.
Consequences of on-target editing of HBG1 and HBG2. (A) Scale diagram of the HBG1 and HBG2 genes with exons shown as rectangles and on-target Cas9:sgRNA-1 DNA cleavage sites indicated by vertical black arrows. The genes differ by a single nucleotide in exon 3 (open arrows). Primer pair If+Ir amplifies a region surrounding the RNP cleavage site of both genes; NGS of this PCR product identified small on-target indels. Larger deletions presumed to arise from simultaneous dsDNA breaks in HBG2 and HBG1, with the loss of the intervening 4.9 kb (dotted line), were indicated by 2 proxy methods. “ΔHBG2” denotes dropout of HBG2 exon 3 based on the single-nucleotide difference between HBG2 and HBG1, determined by high-throughput sequencing of PCR products generated by a single primer pair (not shown). “Δ366+” denotes a quantitative TaqMan PCR assay that detects deletions ≥366 nt upstream of the Cas9:sgRNA-1 cleavage site in HBG1. (B) Large deletions detected by the ΔHBG2 and Δ366+ assays in CD34+ cells 4 days after editing (Pre-BMT) and 17 weeks after xenotransplantation (Post-BMT). Data reflect studies from 2 different CD34+ cell donors (blue or red dots), with each dot after bone marrow transplantation representing a single mouse. (C) Mutational analysis and %HbF in single BFU-E colonies. Each dot represents a BFU-E colony grouped by number of 4.9-kb deletions (Del) using the ΔHBG2 assay as proxy and the number of indels in the remaining BCL11A binding motifs of HBG2, HBG1, or the modified HBG1 fusion gene formed by deletion repair. Thus, heterozygosity for the 4.9-kb deletion leaves 3 potential remaining BCL11A-binding motifs and associated HBG coding regions, whereas homozygosity leaves only 2 such motifs. The bars represent the percentages of HbF as the means ± SDs. **P < .01, ***P < .001, ****P < .0001 (by unpaired Student t test). (D) Inversions resulting from simultaneous editing of HBG2 and HBG1 target sites were characterized by PCR analysis using the indicated primer pairs. The agarose gel image shows PCR products from the bulk-edited HUDEP-2 (H2) erythroid cell line and CD34+ cells. ns, not significant.
To measure large indels clonally and investigate whether the associated loss of HBG2 impaired HbF production, we analyzed burst-forming unit erythroid (BFU-E) colonies from edited and control CD34+ cells (Figure 4C). The frequency of hematopoietic colonies was decreased by ∼30% in CD34+ cells electroporated with Cas9:sgRNA-1 RNP vs Cas9 alone (supplemental Figure 4A), with the proportions of myeloid and erythroid colonies being similar (supplemental Figure 4B). In pools of ∼1000 gene-edited colonies, the small indel frequency was 57.3% (supplemental Figure 4C) and the %HbF increased from 8.1% ± 0.6% in controls to 27.7% ± 2.5% after editing (supplemental Figure 4D). In 124 BFU-E colonies analyzed individually, the frequency of small on-target indels was 45%, and the frequency of large indels measured by loss of HBG2 exon 3 was 24% (Figure 4C). Colonies with no on-target edits contained 9.5% ± 5.8% HbF (n = 47). Most edited colonies contained either 4 γ-globin alleles, all with small indels that disrupted BCL11A binding motifs (n = 15; HbF, 48.4% ± 30%), or 1 deleted HBG2 allele with the BCL11A-binding motifs disrupted in the 3 remaining γ-globin genes (n = 28; HbF, 40.8% ± 17%). Thus, the loss of a single HBG2 allele, presumably reflecting the 4.9-kb deletion, did not appreciably alter the %HbF.
Simultaneous dsDNA breaks at the HBG1 and HBG2 promoters can also cause inversion of the intervening DNA (Figure 4D), predicted to eliminate the expression of both genes. Using primer pairs that detect this inversion, we identified multiple PCR products specifically in edited HUDEP-2 cells and CD34+ HSPCs. Sanger sequencing showed that these inverted segments contained interrupted sequences and deletions ranging from 236 to 1916 bp. We detected the inversion in 1 of 96 BFU-E colonies (1%) generated from bulk-edited CD34+ HSPCs with an overall on-target small indel frequency of 84%. Thus, this inversion likely occurs at low rates.
Cas9:sgRNA-1 RNP cleaves DNA specifically
We characterized the genome-wide activity of Cas9:sgRNA-1 by incubating RNP with purified genomic DNA followed by circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq), a sensitive in vitro method for enriching for Cas9-cleaved genomic DNA and identifying the breakpoints by NGS.31 In 3 experimental replicates, we consistently detected 26 potential off-target candidate sites (Figure 5, left panel). However, no mutations were detected at these sites by NGS of PCR products generated from edited HSPCs (sensitivity 0.1%-0.01%) (Figure 5, right panel).
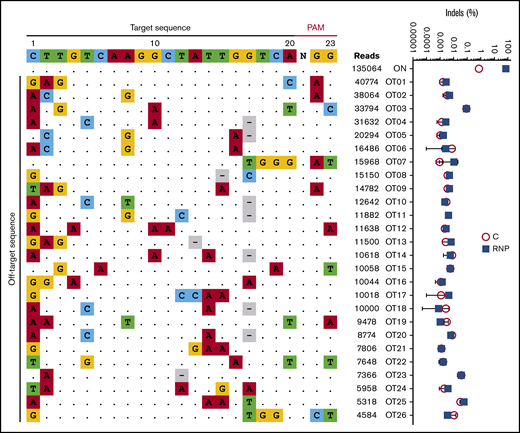
No off-target mutations are detected by a sensitive assay in CD34+HSPCs after editing at the γ-globin gene promoters. Purified genomic DNA from normal CD34+ cell–derived erythroblasts was analyzed by the CIRCLE-seq method for defining Cas9 genome-wide activity in vitro. The left panel shows the 25 most frequent candidate off-target sites for Cas9:sgRNA-1 RNP identified by CIRCLE-seq analysis on purified genomic DNA. The on-target sites including sgRNA and PAM sequences are shown at the top. Off-target sites are ordered by CIRCLE-seq read count, with matches to the intended target site shown as dots and mismatches shown as colored nucleotides. The right panel shows the indel frequencies at candidate off-target sites validated by targeted high-throughput sequencing of CD34+ HSPCs edited with Cas9:sgRNA-1 RNP using the Neon Transfection System (blue squares) and control nonedited CD34+ cells (red circles). The threshold of detection for NGS is ∼0.01% to 0.1%.
No off-target mutations are detected by a sensitive assay in CD34+HSPCs after editing at the γ-globin gene promoters. Purified genomic DNA from normal CD34+ cell–derived erythroblasts was analyzed by the CIRCLE-seq method for defining Cas9 genome-wide activity in vitro. The left panel shows the 25 most frequent candidate off-target sites for Cas9:sgRNA-1 RNP identified by CIRCLE-seq analysis on purified genomic DNA. The on-target sites including sgRNA and PAM sequences are shown at the top. Off-target sites are ordered by CIRCLE-seq read count, with matches to the intended target site shown as dots and mismatches shown as colored nucleotides. The right panel shows the indel frequencies at candidate off-target sites validated by targeted high-throughput sequencing of CD34+ HSPCs edited with Cas9:sgRNA-1 RNP using the Neon Transfection System (blue squares) and control nonedited CD34+ cells (red circles). The threshold of detection for NGS is ∼0.01% to 0.1%.
At ≥16 weeks after xenotransplantation, gene-edited human donor cells contained no chromosomal rearrangements detected by G-band karyotyping (supplemental Figure 5) or fluorescence in situ hybridization (FISH) with a probe located distal to the γ-globin gene loci on human chromosome 11 (supplemental Figure 6). Overall, our data suggest that Cas9:sgRNA-1 RNP targets the HBG1 and HBG2 promoters with high specificity and without detectable off-target DNA mutations at the sites analyzed.
Genome editing of HSCs from patients with SCD induces HbF expression in erythroid progeny
We next analyzed the effects of Cas9:sgRNA-1 gene editing in CD34+ HSPCs from 2 individuals with SCD. As granulocyte colony-stimulating factor (G-CSF) is contraindicated in SCD,32,33 we edited plerixafor-mobilized peripheral blood CD34+ cells. After xenotransplantation into NBSGW mice, edited and control (nonedited) CD34+ cells populated the bone marrow similarly (Figure 6A) and gave rise to similar fractions of human T, B, myeloid, and erythroid cells (Figure 6B). For SCD donor 1, the small on-target indel frequency was 79% before transplantation and remained high at 16 weeks after transplantation (72% ± 3%) (Figure 6C). For SCD donor 2, the indel frequency was 55% before transplantation and declined to 18% ± 2% after transplantation. The spectrum of on-target indels in SCD CD34+ HSPCs was similar to that observed after editing normal G-CSF–mobilized CD34+ HSPCs and did not change appreciably after xenotransplantation (supplemental Figure 7).
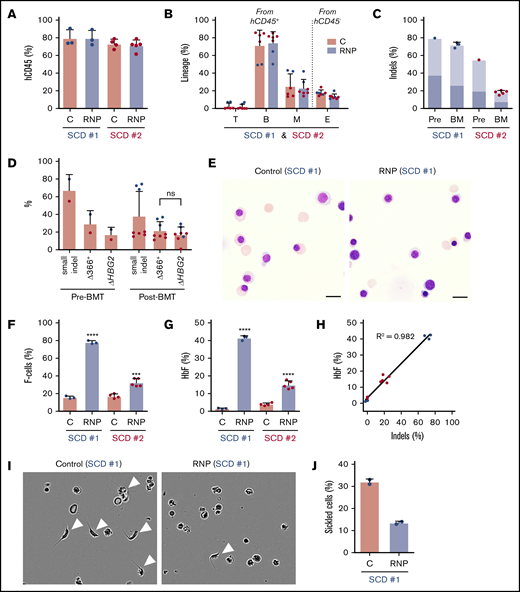
HBG1 and HBG2 promoters in CD34+HSPCs from individuals with SCD induces HbF expression in erythroid progeny generated after xenotransplantation. Plerixafor-mobilized CD34+ cells from 2 individuals with SCD (represented by blue and red) were edited with Cas9:sgRNA-1 RNP by using the Neon Transfection System. They were then transplanted into NBSGW mice via tail-vein injection. Donor-cell controls (C) were processed in parallel, but not electroporated. Donor cells were analyzed in recipient bone marrow at 16 to 18 weeks after transplantation. (A) Normalized human cell chimerism (hCD45%) in bone marrow for the 2 SCD donors. (B) Chimerism for specific human lineages in recipient bone marrow, labeled as in Figure 3B. (C) The indel fraction in CD34+ cells at 4 days after gene editing (Pre) and in bone marrow after xenotransplantation. Dark blue represents the fraction of indels with the 13-nt HPFH deletion; light blue represents all other indels. (D) Large deletions were quantified by the ΔHBG2 and Δ366+ assays (Figure 4A) in edited CD34+ cells before 16 weeks after transplantation. (E) Morphology of human CD235a+ erythroblasts in bone marrow of mice transplanted with control or RNP-edited CD34+ cells. The images were acquired using a Nikon Eclipse Ni microscope with a 60× objective and Nikon NIS-Elements software (scale bars, 10 μm). (F) The F-cell fraction in bone marrow CD235a+ erythroblasts. (G) The %HbF in CD235a+ erythroblasts isolated from recipient bone marrow, as determined by ion-exchange HPLC. (H) The correlation of the %indels with the %HbF in CD235a+ erythroblasts isolated from recipient bone marrow. (I) Human erythroblasts isolated from mouse recipient bone marrow were incubated for 8 hours in 2% O2 and visualized by phase-contrast microscopy using the IncuCyte S3 Live-Cell Analysis System (Sartorius) with a 20× objective. The white arrowheads indicate cells with sickle-like morphology. (J) The sickled cell fraction after hypoxia in edited and control cells was determined by analyzing micrographs such as those in panel I. More than 300 cells in 2 independent experiments were scored by 2 blinded observers. All bar charts show data as the mean ± SD. Each dot represents a single recipient mouse from donor 1 (blue) or 2 (red). ****P < .0001, ***P < .001, **P < .01, and *P < .05 (by unpaired Student t test).
HBG1 and HBG2 promoters in CD34+HSPCs from individuals with SCD induces HbF expression in erythroid progeny generated after xenotransplantation. Plerixafor-mobilized CD34+ cells from 2 individuals with SCD (represented by blue and red) were edited with Cas9:sgRNA-1 RNP by using the Neon Transfection System. They were then transplanted into NBSGW mice via tail-vein injection. Donor-cell controls (C) were processed in parallel, but not electroporated. Donor cells were analyzed in recipient bone marrow at 16 to 18 weeks after transplantation. (A) Normalized human cell chimerism (hCD45%) in bone marrow for the 2 SCD donors. (B) Chimerism for specific human lineages in recipient bone marrow, labeled as in Figure 3B. (C) The indel fraction in CD34+ cells at 4 days after gene editing (Pre) and in bone marrow after xenotransplantation. Dark blue represents the fraction of indels with the 13-nt HPFH deletion; light blue represents all other indels. (D) Large deletions were quantified by the ΔHBG2 and Δ366+ assays (Figure 4A) in edited CD34+ cells before 16 weeks after transplantation. (E) Morphology of human CD235a+ erythroblasts in bone marrow of mice transplanted with control or RNP-edited CD34+ cells. The images were acquired using a Nikon Eclipse Ni microscope with a 60× objective and Nikon NIS-Elements software (scale bars, 10 μm). (F) The F-cell fraction in bone marrow CD235a+ erythroblasts. (G) The %HbF in CD235a+ erythroblasts isolated from recipient bone marrow, as determined by ion-exchange HPLC. (H) The correlation of the %indels with the %HbF in CD235a+ erythroblasts isolated from recipient bone marrow. (I) Human erythroblasts isolated from mouse recipient bone marrow were incubated for 8 hours in 2% O2 and visualized by phase-contrast microscopy using the IncuCyte S3 Live-Cell Analysis System (Sartorius) with a 20× objective. The white arrowheads indicate cells with sickle-like morphology. (J) The sickled cell fraction after hypoxia in edited and control cells was determined by analyzing micrographs such as those in panel I. More than 300 cells in 2 independent experiments were scored by 2 blinded observers. All bar charts show data as the mean ± SD. Each dot represents a single recipient mouse from donor 1 (blue) or 2 (red). ****P < .0001, ***P < .001, **P < .01, and *P < .05 (by unpaired Student t test).
The frequency of 4.9-kb HBG2-HBG1 intergenic deletions in edited SCD donor 1 CD34+ cells detected by the ΔHBG2 and Δ366+ assays (Figure 4A) was ∼30% before transplantation and 33% after transplantation (Figure 6D). These large deletions were less frequent in edited SCD donor 2 CD34+ cells, consistent with relatively low frequencies of small indels. At 16 weeks after xenotransplantation with edited SCD donor 1 HSPCs, we purified donor CD34+ cells and analyzed them by FISH using a probe for the HBG2-HBG1 intergenic region and a control probe for an adjacent downstream region including the HBB gene (supplemental Figure 8A). Most cells derived from gene-edited donor HSPCs (42 of 75 cells analyzed) were heterozygous for the 4.9-kb HBG2-HBG1 intergenic deletion (overall allele frequency, 32%) (supplemental Figure 8B).
At 16 weeks after transplantation, bone marrow erythroblasts derived from gene-edited and control CD34+ HSPCs exhibited similar morphologies (Figure 6E) and maturation profiles as determined by flow cytometry (supplemental Figure 9A-B). Erythroblasts derived from gene-edited HSPCs from SCD patient 1 CD34+ cells contained 77.8% ± 1.9% F cells as compared with 15.7% ± 1.6% in control nonedited erythroblasts (Figure 6F; supplemental Figure 9C). The %HbF was 42.9% ± 1.5% after gene editing vs 1.3% ± 0.5% in control erythroblasts (Figure 6G). Erythroblasts generated in vivo from HSPCs from SCD patient 2 also exhibited substantially increased HbF induction after undergoing gene editing, despite lower on-target indel frequencies (Figure 6C,F-G). HbF induction in SCD erythroid cells correlated with indel formation (Figure 6H), similar to that which we observed in erythroid cells derived from normal donor HSPCs (Figure 3K). To investigate the potential therapeutic benefits of HbF induction, we used immunomagnetic beads to purify CD235a+ erythroid cells from the bone marrow of mice that had been transplanted with HSPCs from SCD patient 1. After being subjected to hypoxia (2% O2) for 8 hours, 32% ± 1.0% of control erythroid cells exhibited sickled morphology as compared with 13.5% ± 1.0% of gene-edited cells (Figure 6I-J). Thus, HbF induction resulting from Cas9:sgRNA-induced disruption of the BCL11A-binding site of the HBG1 and HBG2 gene promoters inhibits erythroid cell sickling.
Superior HSC gene editing by Cas9-3xNLS
Our findings demonstrate that genome editing of the HBG1 and HBG2 promoters in HSCs induces HbF in erythroid progeny. However, on-target indel frequencies varied between CD34+ cell donors and tended to decline over time after xenotransplantation (Figures 3C and 6C). The Streptococcus pyogenes Cas9 protein used in our studies described thus far contains 2 tandem C-terminal SV40 nuclear-localization signals (NLSs; Cas9-2xNLS).34 We recently reported improved gene editing of human HSPCs using a modified SpCas9 containing a c-Myc–like NLS at the N terminus and both SV40 and nucleoplasmin NLSs at the C terminus (Cas9-3xNLS) (Figure 7A).35
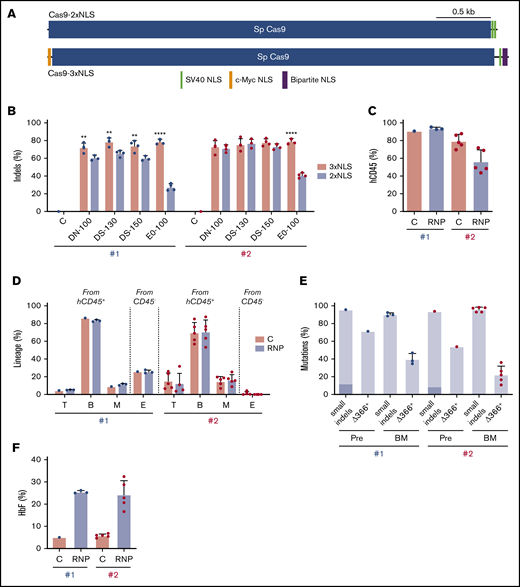
Improved genome editing of the HBG1 and HBG2 promoters in HSCs by Cas9-3xNLS. (A) Schematic overview of Cas9-2xNLS and Cas9-3xNLS proteins showing locations of nuclear localization signals. (B) Normal human CD34+ cells from 2 different donors (#1 and #2 represented by blue and red dots, respectively) were edited using 4 different delivery methods with the Lonza 4D-Nucleofector platform (x-axis) with RNPs composed of sgRNA-1 and either Cas9-3xNLS (red bar) or Cas9-2xNLS (blue bar), then cultured in CD34+ cell maintenance medium (supplemental Table 2). Nonedited donor-cell controls (C) were processed in parallel, but not electroporated. Indels were analyzed at day 3 by NGS. (C-F) Normal CD34+ cells from 2 different donors were edited with the Lonza 4D-Nucleofector (EO-100 program) and RNP composed of sgRNA-1:Cas9-3xNL and then transplanted into NBSGW mice. (C) Normalized chimerism of human CD45+ donor cells in recipient bone marrow at 16 weeks after xenotransplantation. (D) Chimerism for different human lineages, labeled as in Figure 3B. (E) Frequencies of small indels detected by TIDE assay and large (4.9 kb) deletions detected by the Δ366+ assay (Figure 4A) in donor CD34+ cells 5 days after editing (Pre) and 16 weeks after bone marrow transplantation. The 13-nt HPFH mutation is indicated in dark blue; all other mutations are represented by light blue. (F) Donor CD34+ cells were purified from recipient bone marrow and grown in culture for 18 days under erythroid differentiation conditions. The bar chart shows %HbF as determined by ion-exchange HPLC. Each dot represents a biological replicate experiment with 2 different CD34+ cell donors (blue and red). **P < .01 and ****P < .0001 (unpaired Student t test).
Improved genome editing of the HBG1 and HBG2 promoters in HSCs by Cas9-3xNLS. (A) Schematic overview of Cas9-2xNLS and Cas9-3xNLS proteins showing locations of nuclear localization signals. (B) Normal human CD34+ cells from 2 different donors (#1 and #2 represented by blue and red dots, respectively) were edited using 4 different delivery methods with the Lonza 4D-Nucleofector platform (x-axis) with RNPs composed of sgRNA-1 and either Cas9-3xNLS (red bar) or Cas9-2xNLS (blue bar), then cultured in CD34+ cell maintenance medium (supplemental Table 2). Nonedited donor-cell controls (C) were processed in parallel, but not electroporated. Indels were analyzed at day 3 by NGS. (C-F) Normal CD34+ cells from 2 different donors were edited with the Lonza 4D-Nucleofector (EO-100 program) and RNP composed of sgRNA-1:Cas9-3xNL and then transplanted into NBSGW mice. (C) Normalized chimerism of human CD45+ donor cells in recipient bone marrow at 16 weeks after xenotransplantation. (D) Chimerism for different human lineages, labeled as in Figure 3B. (E) Frequencies of small indels detected by TIDE assay and large (4.9 kb) deletions detected by the Δ366+ assay (Figure 4A) in donor CD34+ cells 5 days after editing (Pre) and 16 weeks after bone marrow transplantation. The 13-nt HPFH mutation is indicated in dark blue; all other mutations are represented by light blue. (F) Donor CD34+ cells were purified from recipient bone marrow and grown in culture for 18 days under erythroid differentiation conditions. The bar chart shows %HbF as determined by ion-exchange HPLC. Each dot represents a biological replicate experiment with 2 different CD34+ cell donors (blue and red). **P < .01 and ****P < .0001 (unpaired Student t test).
To test whether Cas9-3xNLS edits HSPCs more consistently, we electroporated RNP complexes containing sgRNA-1 and either Cas9-2xNLS or Cas9-3xNLS into normal human CD34+ cells with the Lonza Nucleofector 4-D platform and then measured the resulting indel frequencies after 3 days of culture in HSC media. Cas9-3xNLS maintained consistently high indel rates (65.9% to 84.2%) across 2 donors and 4 different delivery conditions, whereas under identical conditions, Cas9-2xNLS/sgRNA-1 RNP generated more variable indel rates, ranging from 24.1% to 82.1% (Figure 7B).
Next, we transfected Cas9-3xNLS/sgRNA-1 RNP into CD34+ HSPCs from 2 normal donors and transplanted the cells into NBSGW mice. At 16 weeks after transplantation, human hematopoietic chimerism in the bone marrow measured by human CD45 expression was 93.6% ± 0.9% for donor 1 and 56.5% ± 5.8% for donor 2 (Figure 7C). This difference may arise from variation in the fraction of repopulating HSCs within the bulk donor CD34+ cell population from different donors. Bone marrow chimerism for T, B, myeloid, and erythroid cell populations was similar for edited and control donor cells, although erythroid chimerism was low for donor 2 (Figure 7D). The indel frequencies measured by TIDE (tracking of indels by decomposition36 ) of donor CD34+ HSPCs were similar before and after transplantation, ranging from 86% to 98%. The frequency of the 4.9-kb intergenic HBG2-HBG1 deletion in CD34+ donor cells at 16 weeks after transplantation estimated by the TaqMan Δ366+ assay was ∼40% for donor 1 and 22% for donor 2 (Figure 7E). We purified donor-derived CD34+ cells from recipient bone marrow and cultured them under erythroid differentiation conditions. The HbF level was 25% ± 5% in RBCs derived from gene-edited donor CD34+ cells but only 6% ± 1% in control RBCs (Figure 7F). Together, the results of these studies indicate that CD34+ HSPCs edited with Cas9-3xNLS/sgRNA-1 RNP maintained consistently high levels of editing after xenotransplantation. Although additional comparisons are required, our experiments suggest that optimized delivery of RNP with Cas9-3xNLS or similar enhanced-activity variants should facilitate robust editing of the HBG1 and HBG2 promoters in long-term engrafting HSCs, with HbF being induced in the RBC progeny to therapeutic levels for SCD and β-thalassemia.
Discussion
Previously, we demonstrated that Cas9-mediated disruption of a repressor element in the HBG1 and HBG2 promoters induces erythroid HbF expression.16 Here, we advance those results by showing that transient expression of Cas9 RNPs in human CD34+ cells results in high-frequency editing of the target site (≤80%) in repopulating HSCs with induction of HbF to potentially therapeutic levels in RBC progeny and no detectable genotoxicity. Editing at this site precisely disrupts a binding motif for the BCL11A transcriptional repressor protein, consistent with findings that most on-target indels activate HBG1/HBG2 transcription.16 Our study generated several important new findings.
First, our methods achieved substantially higher rates of target-site editing in HSCs than reported previously after transient expression of Cas9 RNPs30 or transcription activator-like effector nucleases,37 which will likely translate to greater therapeutic benefits. The variability of HSC editing that we observed in our studies using Cas9-2xNLS could be due to differences in donors, CD34+ mobilization methods (ie, G-CSF vs plerixafor), or CD34+ cell purification. Notably, the use of chemically modified sgRNA and Cas9-3xNLS35 achieved consistently high editing activity across donors and delivery methods, in contrast to a commonly used Cas9 with 2 SV40 NLSs fused to the C terminus (Cas9-2xNLS). The improvement in editing by Cas9-3xNLS may arise from NLS signals at both ends of the protein and/or the use of 3 unique NLS signals that interact synergistically with nuclear import machinery.38
Second, our study demonstrates high-frequency editing (≤70%) of plerixafor-mobilized CD34+ HSPCs from individuals with SCD, which represents the cell product most likely to be manipulated therapeutically. Increased HbF levels enhance the survival of erythroblasts and mature RBCs in patients with β-thalassemia or SCD.39-46 The therapeutic benefits of our strategy depend on the fraction of repopulating HSCs edited and the resultant levels and cellular distribution of HbF. Because circulating human RBCs are cleared very rapidly in mice, we are unable to measure the survival advantage conferred by gene editing. After allogeneic bone marrow transplantation for SCD, ∼20% donor HSC chimerism can result in 100% circulating donor RBCs and marked clinical improvement.47,48 In natural history studies, pancellular HbF levels >20% to 30% are suggested to protect against SCD morbidities, consistent with biochemical studies of HbS polymerization.49-51 Based on these parameters extrapolated from clinical studies, the preclinical outcomes achieved in this study (70% to 80% indel rate in HSCs, with most or all RBC progeny expressing HbF at an average level of 30% to 40%) are likely to produce clinical benefits. One limitation of our study is that on-target editing frequencies in HSCs varied considerably between experiments and donors in initial experiments. However, our data using newly developed Cas9-3xNLS suggest that consistent high-level editing of the HBG1/HBG2 promoters in repopulating HSCs can now be achieved, similar to what was recently reported for an erythroid-specific enhancer in the BCL11A gene.35
Third, our study provides critical new preclinical safety assessments by showing that the 4.9-kb deletion resulting from simultaneous on-target DSBs in HBG1 and HBG2 promoters is not deleterious to HSCs or RBC precursors. Analysis of clonal BFU-E colonies showed that HbF expression was not impaired by deletion of HBG2. In most clones harboring the 4.9-kb deletion, the BCL11A-binding motif was destroyed at the DSB repair site, thereby favoring expression of the reconstituted HBG1 gene. Expression of β-like globin genes is facilitated via interaction with an upstream enhancer, termed locus control region.52 Loss of HBG2 may eliminate competition for the locus control region, thereby favoring upregulation of the remaining HBG1 gene. Interestingly, a germline variant similar to the 4.9-kb deletion noted in our study has been identified in multiple unrelated individuals. This variant arises from unequal crossing over between microsatellite markers in the second introns of HBG2 and HBG1, resulting in a 5-kb deletion that generates a single chimeric γ-globin gene with exons 1 and 2 originating from HBG2 and exon 3 from HBG1.53,54 Individuals with this variant have RBCs with reduced Gγ to Aγ ratio as infants but are otherwise normal throughout life. These clinical observations, along with our experimental data, suggest that the 4.9-kb deletion arising from our gene-editing strategy will not have deleterious effects on globin expression, erythroid development, or RBC physiology.
Additionally, our studies did not detect off-target DSBs in edited HSCs at top candidate sites identified by CIRCLE-seq, and no chromosomal abnormalities were detected by G-banding or FISH analysis of >200 CD34+ HSPCs after xenotransplantation. For ultimate therapeutic application, it will be important to examine all sites reproducibly identified by CIRCLE-seq after editing the HBG promoters with Cas9-3xNLS and perform further assessments for structural variants induced by our gene-editing protocol.
In summary, our studies demonstrate that it is possible to achieve high-level editing of the HBG1/HBG2 promoters in G-CSF–mobilized CD34+ cells from normal individuals and plerixafor-mobilized CD34+ cells from individuals with SCD, that editing does not impair hematopoiesis, and that productive editing results in HbF induction to potentially therapeutic levels, with no off-target DSBs or chromosomal rearrangements being detected. These preclinical safety and efficacy studies support a promising strategy for modifying human HSPCs to treat human β-hemoglobinopathies.
Acknowledgments
The authors thank the following core facilities and individuals at St. Jude Children’s Research Hospital: Flow Cytometry (Richard Ashmun, Deanna Langfitt, and Stacie Woolard), Animal Resource Center (Chandra Savage), and Veterinary Pathology (Patricia J. Varner).
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant P01 HL053749 (D.E.B., S.P.-M., S.Q.T., and M.J.W.), Doris Duke Charitable Foundation grant DDCF-2017093 (S.Q.T. and M.J.W.), National Institutes of Health, National Heart, Lung, and Blood Institute grant U01HL145793 (S.Q.T.), National Institute of Allergy and Infectious Diseases R01AI117839, and National Institute of General Medical Sciences R01GM115911 (S.A.W.), The Assisi Foundation of Memphis (M.J.W.), and St. Jude/ALSAC and the St. Jude Collaborative Research Consortium, “Novel Gene Therapies for Sickle Cell Disease." The St. Jude Cytogenetic Shared Resource Laboratory is supported by National Institutes of Health, National Cancer Institute grant P30 CA21765 and American Lebanese Syrian Associated Charities.
Authorship
Contribution: J.-Y.M. contributed to study design, experiments, analyses, and drafting of the manuscript; P.A.D. contributed to experiments, analyses, and drafting of the manuscript; T.M. and V.K. contributed to experiments, data analysis, and drafting of manuscript; S.C.F. contributed to experiments and mouse maintenance; M.M.H., K.L., M.D.N., N.U., and S.A.W. contributed reagents; S.K., S.N.P., S.S.P., D.S., K.J.W., J.Z., Y.W., and Y.Y. contributed to experiments; C.R.L. contributed to experiments and data analysis; S.T.P. contributed to data analysis; B.Y.R., A.S., and J.F.T. contributed to study design and contributed reagents; D.E.B. and S.P.-M. contributed to study design; and S.Q.T. and M.J.W. contributed to study design, data analysis, and drafting of the manuscript.
Conflict-of-interest disclosure: M.J.W. is a consultant for GlaxoSmithKline, Rubius Inc., Cellarity Inc., Beam Therapeutics, and Esperion; none of the consulting work is relevant to the current project. The remaining authors declare no competing financial interests.
Correspondence: Mitchell J. Weiss, Department of Hematology, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, Memphis, TN 38105; e-mail: mitch.weiss@stjude.org; and Shengdar Q. Tsai, Department of Hematology, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl, Memphis, TN 38105; e-mail: shengdar.tsai@stjude.org.

