

Key Points
DNA replication timing of >100 pediatric leukemic samples identified BCP-ALL subtype-specific genome alteration signatures.
Comparative analyses identified features of specific stages of B-cell differentiation and potential associations with clinical outcome.

Abstract
Human B-cell precursor acute lymphoid leukemias (BCP-ALLs) comprise a group of genetically and clinically distinct disease entities with features of differentiation arrest at known stages of normal B-lineage differentiation. We previously showed that BCP-ALL cells display unique and clonally heritable, stable DNA replication timing (RT) programs (ie, programs describing the variable order of replication and subnuclear 3D architecture of megabase-scale chromosomal units of DNA in different cell types). To determine the extent to which BCP-ALL RT programs mirror or deviate from specific stages of normal human B-cell differentiation, we transplanted immunodeficient mice with quiescent normal human CD34+ cord blood cells and obtained RT signatures of the regenerating B-lineage populations. We then compared these with RT signatures for leukemic cells from a large cohort of BCP-ALL patients with varied genetic subtypes and outcomes. The results identify BCP-ALL subtype-specific features that resemble specific stages of B-cell differentiation and features that seem to be associated with relapse. These results suggest that the genesis of BCP-ALL involves alterations in RT that reflect biologically significant and potentially clinically relevant leukemia-specific epigenetic changes.
Introduction
DNA replication timing (RT) refers to the temporal order in which defined units of chromosomes replicate during the course of S phase. The regulatory units of RT correspond to units of structural organization and are organized into higher-order 3D spatial compartments in the nucleus that replicate at distinct times during S phase.1,2 Changes in RT affect at least half the genome during normal development and differentiation,1,3,4 and RT profiles are characteristic of a given cell type.5-8 Early RT correlates with transcriptional activity, but there are many exceptions,9,10 and RT signatures can identify differences between diseased and normal tissue that are not identified by standard transcriptome analyses.11,12 RT signatures may therefore provide a novel genre of clinical biomarkers that reflect large-scale genome architecture. We previously described disease- and patient-specific features in the RT profiles of B-cell precursor acute lymphoid leukemia (BCP-ALL) cells2,13 and showed that they remained stable in serially passed patient-derived xenografts in immunodeficient mice.14 Here, we investigated the biological relevance of RT alterations to BCP-ALL by examining the relationship of BCP-ALL RT profiles to specific stages of normal B-cell differentiation from which this class of leukemias derive and their potential prognostic significance. Results establish the existence of leukemia-specific RT signatures that suggest previously unknown associations with specific BCP-ALL subtypes and their responses to therapy.
Methods
Patient samples
Primary BCP-ALL patient samples were obtained with informed consent according to protocols approved by the Institutional Review Board of the Oregon Health & Science University and St. Jude Children’s Research Hospital. Mononuclear cells were obtained from bone marrow aspirates by Ficoll density gradient centrifugation, and viably frozen cells were stored in 90% fetal bovine serum (FBS) and 10% dimethyl sulfoxide.
Normal cells
Human cord blood (CB) samples were obtained with informed consent, anonymized, and used according to procedures approved by the Research Ethics Board of the University of British Columbia. Low-density CD3–CD19–CD11b– cells depleted of neutrophils and red blood cells were isolated on Lymphoprep using RosetteSep, and the >90% pure CD34+ cells were isolated using EasySep (STEMCELL Technologies). Cells were stored frozen at −176°C in dimethyl sulfoxide with 90% FBS. Before transplanting the cells into mice, they were thawed in Iscove modified Dulbecco medium with 10% FBS (STEMCELL Technologies) and 10 mg/mL DNase I (Sigma Aldrich), centrifuged, and resuspended in Hanks balanced salt solution (STEMCELL Technologies) with 2% FBS.
Xenografts
Two × 104 to 10 × 104 normal human CD34+ CB cells (2 biological replicates consisting of pooled CB cells from 3 individuals) were IV injected into 8- to 12-week-old adult female NRG mice within a few hours of being exposed to 8.5 cGy of 137Cs γ-rays delivered over 3 hours. Mice were bred in the Animal Resource Centre of the British Columbia Cancer Research Centre and treated using procedures approved by the Animal Care Committee of the University of British Columbia. Ten to 15 weeks later, pelvic, femoral, and tibial bone marrow and spleen cells were isolated and sorted for subsets by fluorescence-activated cell sorting (FACS).15
Simultaneous sorting for surface markers and DAPI
Red blood cell lysis was performed by using ammonium chloride solution (STEMCELL Technologies) for 10 minutes on ice. A crude enrichment for human cells was performed using an EasySep Mouse/Human Chimera Isolation Kit (STEMCELL Technologies). Cells were spun down and resuspended in Iscove modified Dulbecco medium supplemented with 10% FBS (both from STEMCELL Technologies) and 100 µM bromodeoxyuridine (BrdU; BD Pharmingen) and placed in a humidified 37°C (5% CO2 in air) environment for 2 hours. Cells were washed once with Dulbecco phosphate-buffered saline and 2% FBS and stained with 1:25 anti-human CD45 fluorescein isothiocyanate (clone 2D1; STEMCELL Technologies), 1:50 anti-human CD45 allophycocyanin-ef780 (clone HI30; eBiosciences), 1:100 anti-human CD34 AlexaFluor 647 (clone 581), 1:50 anti-human CD19 phycoerythrin-Cy7 (clone HIB19), and 1:800 anti-human ROR1 phycoerythrin (clone 2A2, all from BioLegend) for 30 minutes on ice. Cells were once again washed, then resuspended in 8 mg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) plus 200 mg/mL Digitonin (Sigma) in phosphate-buffered saline plus 2% FBS. Cells were sorted on a BD FACSAria Fusion sorter directly into QIAGEN Buffer AL for downstream processing.
Repli-ChIP, Repli-seq, and RT signatures
Repli-capture-seq
Roche SeqCap EZ Developer Library (cat #06471684001; IRN/Design Name:4000016400) was designed to tile a 250-bp capture region within the central 4-kb target region from each 10-kb window of hg19 avoiding nonspecific sequences so that the maximum distance between 2 capture regions was 14 kb, with a 6-kb minimum distance between 2 capture regions. For Repli-capture-seq, G1 and S phase total genomic DNA libraries (average 150-bp insert size) were made from cells isolated by FACS from each patient sample using NEBNext Ultra DNA Library Prep Kit (NEB E7370; Illumina) and single indexed using NEB E7335 or NEB E7500. Up to 12 indexed libraries were pooled, and representative 250-bp regions from each 10-kb window throughout the genome were captured by using the Roche SeqCap EZ Developer Library (cat #06471684001; IRN/Design Name:4000016400), the SeqCap EZ Pure Capture Bead Kit (cat #06 977 952 001), and the SEQCAP EZ Reagent Kit Plus (cat #06 953 212 001), according to the manufacturer’s instructions. After capture, target enrichment was confirmed by quantitative polymerase chain reaction. Target regions were enriched 51 to 221 times compared with the precapture library, and nontarget regions were reduced by 0.02 to 0.16 times.
RT profiles from deep WGS
Paired-end whole-genome sequencing (WGS) on diagnostic leukemic blasts and matched germ line DNA samples from TCF3-PBX1 patients treated at St. Jude Children’s Research Hospital was performed using the Illumina sequencing platform (Illumina Inc., San Diego, CA). The leukemic genomes had an average haploid coverage of 28×. RT was inferred from DNA copy number as previously described.20
TCF3-PBX1 siRNA transfection
Cell lines were transfected using the Lonza Amaxa Nucleofector kit CA 137 program with 6 μg each of 2 separate E2A-PBX1–specific short interfering RNAs (siRNAs) (Eurofins/Operon): CUC CUA CAG UGU UUU GAG U and CAG UGU UUU GAG UAU CCG. These siRNAs were tagged at the 3′end with Cyanine-3 NHS Ester (Cy-3) and Cyanine-5 NHS Ester (Cy-5), respectively, to monitor transfection efficiency by fluorescence microscopy.
Data sharing
Data from the RT data set are available at the National Center for Biotechnology Information Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) under series number GSE130374.
Results
Normal human hematopoietic cell types display distinct RT signatures
We transplanted normal human CD34+ CB cells into sublethally irradiated nonobese diabetic Rag1−/−IL2Rγc−/− (NRG) mice and isolated human CD34+CD38−, CD34+CD19+, CD34−CD19+, and total CD34+ (mixed progenitors) cells by FACS from their bone marrow and spleens 2 to 3 months later (Figure 1A). Biological replicates consisted of cells regenerated from different pooled CB cells. The CD34+CD19+ and CD34−CD19+ cells obtained in 1 of the replicate experiments were also sorted for the presence or absence of ROR1 expression. As expected,21 CD34+CD19+ROR1+ cells were rare, which allowed purification of only 300 cells from the early and late S phase fractions, simultaneously sorted for surface marker expression and DNA content, from which good-quality Repli-seq profiles were then also obtained.
![RT changes during normal B-lineage differentiation. (A) CD34+ CB cells were transplanted into immunodeficient mice and subsequently regenerated. B-lymphoid differentiation intermediates were isolated by FACS using the cell surface markers shown with associated approximated temporal expression patterns. (B) RT profiling method. (C) Exemplary RT profiles highlighting (pink) differences between immature (red) and mature (blue) B cells. (D) Unsupervised clustering analysis of RT-variable regions identified specific RT signatures (labeled in gray boxes). Dendrogram at the top shows the correlation of all samples based on the 10 350 RT-variable 50-kb segments identified. The heat map shows the RT ratios [= log2(early/late)]. (E) Early human hematopoiesis transcriptome data22 was used to measure gene expression patterns within each RT signature. RT signatures from panel D contained 274 genes with transcriptional patterns coordinated with their RT program. (F) Heatmap of expression patterns for known hematopoietic regulators within the RT signatures from panel D. Exemplary genes for each RT signature are shown. CM, common myeloid progenitors; EP, early progenitors (CD34+CD38–); GM, granulocyte-monocyte progenitors; LP, late progenitors (CD34–CD19+); M, myeloid-erythroid progenitors; ME, megakaryocytic-erythroid progenitors; MP, middle progenitors (CD34+CD19+); Prog, progenitor; T, normal T cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019000641/5/m_advancesadv2019000641f1.png?Expires=1771025763&Signature=4eQ4woCXYvP3FarBBaM9UaxTrhl2Y940p6HVik71gyP~ptHMNV77m0~XejxAD2gGcFwJFfzuBUonwV1Fp8orwG3QZR3rl6N6hZIR7mHZc90bdjWQqeJkY97hIENmDpurjjDO9oUwObxj6G7bCQCuKWsFbRd6mIWW1U7SEvHJE2bD0xAvNS0pYZHrfm0toLoqIfW7Tb1mCIXLSAhaeG87~MLi2arAWLiP4ZZ1ymi48665-pxzH1NfD~1hqZjlh5JFTZ0A7vNRIc0XzHOiP-2eHym~b66nUlhW0krXtFJHfEJJ~upg5lXvZwkYU0ofypsUYT9qimz2CgjEYAKhk4Z-ZA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
RT changes during normal B-lineage differentiation. (A) CD34+ CB cells were transplanted into immunodeficient mice and subsequently regenerated. B-lymphoid differentiation intermediates were isolated by FACS using the cell surface markers shown with associated approximated temporal expression patterns. (B) RT profiling method. (C) Exemplary RT profiles highlighting (pink) differences between immature (red) and mature (blue) B cells. (D) Unsupervised clustering analysis of RT-variable regions identified specific RT signatures (labeled in gray boxes). Dendrogram at the top shows the correlation of all samples based on the 10 350 RT-variable 50-kb segments identified. The heat map shows the RT ratios [= log2(early/late)]. (E) Early human hematopoiesis transcriptome data22 was used to measure gene expression patterns within each RT signature. RT signatures from panel D contained 274 genes with transcriptional patterns coordinated with their RT program. (F) Heatmap of expression patterns for known hematopoietic regulators within the RT signatures from panel D. Exemplary genes for each RT signature are shown. CM, common myeloid progenitors; EP, early progenitors (CD34+CD38–); GM, granulocyte-monocyte progenitors; LP, late progenitors (CD34–CD19+); M, myeloid-erythroid progenitors; ME, megakaryocytic-erythroid progenitors; MP, middle progenitors (CD34+CD19+); Prog, progenitor; T, normal T cells.
RT changes during normal B-lineage differentiation. (A) CD34+ CB cells were transplanted into immunodeficient mice and subsequently regenerated. B-lymphoid differentiation intermediates were isolated by FACS using the cell surface markers shown with associated approximated temporal expression patterns. (B) RT profiling method. (C) Exemplary RT profiles highlighting (pink) differences between immature (red) and mature (blue) B cells. (D) Unsupervised clustering analysis of RT-variable regions identified specific RT signatures (labeled in gray boxes). Dendrogram at the top shows the correlation of all samples based on the 10 350 RT-variable 50-kb segments identified. The heat map shows the RT ratios [= log2(early/late)]. (E) Early human hematopoiesis transcriptome data22 was used to measure gene expression patterns within each RT signature. RT signatures from panel D contained 274 genes with transcriptional patterns coordinated with their RT program. (F) Heatmap of expression patterns for known hematopoietic regulators within the RT signatures from panel D. Exemplary genes for each RT signature are shown. CM, common myeloid progenitors; EP, early progenitors (CD34+CD38–); GM, granulocyte-monocyte progenitors; LP, late progenitors (CD34–CD19+); M, myeloid-erythroid progenitors; ME, megakaryocytic-erythroid progenitors; MP, middle progenitors (CD34+CD19+); Prog, progenitor; T, normal T cells.
RT profiles were generated by Repli-ChIP16 or Repli-seq17 as described in “Methods” and illustrated in Figure 1B-C. RT signatures specific to differentiating B cells were derived by comparisons with similarly generated RT profiles obtained for other normal human hematopoietic cells, including our previously reported data for granulopoietic and erythroid cells generated from adult mobilized peripheral blood cells, stimulated adult T cells, and a series of Epstein Barr virus–immortalized B-cell lines7 with a stringent quality control cutoff.16,17
We then derived RT signatures by using a pipeline that identifies regions of the genome that replicate at times unique to each of 26 normal human cell types.7 In brief, after removing reads from the sex chromosomes, remaining mappable reads for each cell type were first divided into 55 940 50-kb segments, each of which was then assigned an RT value (supplemental Figure 1A). We then applied an unsupervised k-means clustering analysis to all 50-kb genomic segments that changed RT from a log2 ratio of ≥0.5 to –0.5 or less (or vice versa)7 to identify 10 350 features (18.5% of the genome) that replicate significantly differently between cell types (P < 2 × 10−16 using Student t tests). Correlation matrix (supplemental Figure 1B) and dendrogram (Figure 1D) analyses of all samples using only these RT-variable regions confirmed the close matching of data from separately analyzed biological replicates (cells generated from different donor pools) and the clustering of samples by hematopoietic cell type to yield 8 RT signatures (Figure 1D; supplemental Figure 1C-D). Interestingly, some RT-variable regions displayed multiple RT switches between sequential early stages of normal human B-cell differentiation. Gene expression changes for the top 10% of highly variable genes derived from published transcriptome data for multiple human hematopoietic cell types22 were generally coordinated with their RT changes (Figure 1D-F). Moreover, multiple known hematopoiesis key regulators were found in each signature: for example, HMGA2, KIT, and HOX genes that are required for self-renewal activity in hematopoietic stem cells23,24 ; CREB5 linked to early precursor’s quiescence; RUNX2 required for hematopoietic stem cell differentiation25 ; and CD79B required for the pro-B/pre-B transition26 (Figure 1D-F; supplemental Table 2). These results indicate that RT features can discriminate phenotypically different hematopoietic cell types.
Identification of BCP-ALL–specific RT signatures
Genome-wide RT profiles were generated on a panel of 97 BCP-ALL patient samples and cell lines, with a small number of pediatric acute myeloid leukemia (AML) and T-cell acute lymphoid leukemia patient samples and cell lines to serve as comparators (supplemental Table 1). Although our standard early/late (E/L) RT method (Figure 2A) performs well on directly analyzed patients samples13 or transplanted mice,14 the numbers of viable cells or cells able to incorporate BrdU that we obtained from many previously frozen banked samples of patients’ cells were very low, consistent with S phase–specific replication fork collapse.27 Therefore, such samples were first isolated as unlabeled S and G1 phase cells by FACS according to their DNA content (Figure 2A). We then used microarray or WGS to obtain the relative copy numbers of each 50-kb sequence in the S and G1 phase cells and derived RT profiles based on the fact that earlier-replicating sequences are higher in copy number than late-replicating sequences in S phase, but are at equal copy number in the G1 phase (Figure 2A; S/G1 Repli-seq).14,28 Because the dynamic range of G1/S data are inherently less than twofold, a large number of sequencing reads is needed to quantify copy numbers (160 million per sample), making it expensive as a routine assay. Thus, we designed a set of capture oligonucleotides spaced evenly throughout the genome (see “Methods”). This significantly reduced the sequence depth required to cover the breadth of the genome (107 mappable reads) and enabled high-resolution RT profiles to be obtained, scaled, and normalized with all other samples (Figure 2B). Clustering analysis and correlation analysis of constitutive regions of the autosomal genome demonstrated a high concordance between replicate data sets (correlation values >0.95) and confirmed a lack of bias from the use of these different methods (Figure 2C; supplemental Figure 2).
![Methods for genome-wide RT profiling. (A) Bone marrow samples were collected from pediatric BCP-ALL patients and analyzed by 1 of the methods shown. Samples with high viability were analyzed by early/late (E/L) RT methods, and samples with poor viability were analyzed either by S/G1 methods or they were expanded in patient-derived xenografts (PDXs) to rejuvenate their viability for analysis by E/L RT methods.14 RT was measured using either E/L or S/G1 methods.14,28 In the E/L method, asynchronously growing cells are pulse-labeled with 5′-bromo-2′-deoxyuridine (BrdU) and separated by FACS into early and late S phase populations. BrdU-substituted DNA is immunoprecipitated with an anti-BrdU antibody and subjected to either microarray analysis (E/L Repli-ChIP16 or NextGen sequencing [E/L Repli-seq17]) to obtain an RT ratio [= log2(early/late)] along each chromosome. For the S/G1 method, unlabeled cells are sorted into S and G1 phase fractions, the relative abundance of genomic sequences is quantified by either microarray (S/G1 Repli-ChIP) or evenly spaced segments of genomic DNA are captured using oligonucleotides and subject to NGS (Repli-capture-seq). (B) Exemplary RT profiles showing the similarity in results using the various methods after appropriate local polynomial smoothing (Loess). Repli-Chip normal B-cell sanples are shown in grey, Repli-Chip B-ALL in orange, Repli-seq normal B-cells in black, Repli-seq B-ALL in red and Repli-Capture-seq in dark red. (C) Hierarchical clustering analysis of all patient data sets based on their genome-wide RT programs confirms that there is no bias from the different methods exploited for RT profiling.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019000641/5/m_advancesadv2019000641f2.png?Expires=1771025763&Signature=KyUBbSb0Io364U8ueasbjSl1om7d6GhBmrQbSYI266E7n781YsUQMEGshO~-3oQHpyD8Y6txblInMaEv8q8abz8QO2fWSAAHMaIiaVZsbhcq4nsCnQQ2kgu-~uupXoGeZfDP8YU0wTp5hAVHdOhqykGG0no6XxpJUv4zVOCElIOLtQnyOWf8ApY-WgNknNIxOIR9MGPIfyweULLqRrBinuCaqzBC1M33KztVIeZrXFZKYgw0qJ-ZOy0hTbrvnaOlGSTh0fEh3sNEXSlgnchylU0cON~6I7iL~XArQ0yqFqyoFd4Rdw7rgOyOXm0o0OmoYp8EjDnOgpzHs9eSRNWRew__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Methods for genome-wide RT profiling. (A) Bone marrow samples were collected from pediatric BCP-ALL patients and analyzed by 1 of the methods shown. Samples with high viability were analyzed by early/late (E/L) RT methods, and samples with poor viability were analyzed either by S/G1 methods or they were expanded in patient-derived xenografts (PDXs) to rejuvenate their viability for analysis by E/L RT methods.14 RT was measured using either E/L or S/G1 methods.14,28 In the E/L method, asynchronously growing cells are pulse-labeled with 5′-bromo-2′-deoxyuridine (BrdU) and separated by FACS into early and late S phase populations. BrdU-substituted DNA is immunoprecipitated with an anti-BrdU antibody and subjected to either microarray analysis (E/L Repli-ChIP16 or NextGen sequencing [E/L Repli-seq17 ]) to obtain an RT ratio [= log2(early/late)] along each chromosome. For the S/G1 method, unlabeled cells are sorted into S and G1 phase fractions, the relative abundance of genomic sequences is quantified by either microarray (S/G1 Repli-ChIP) or evenly spaced segments of genomic DNA are captured using oligonucleotides and subject to NGS (Repli-capture-seq). (B) Exemplary RT profiles showing the similarity in results using the various methods after appropriate local polynomial smoothing (Loess). Repli-Chip normal B-cell sanples are shown in grey, Repli-Chip B-ALL in orange, Repli-seq normal B-cells in black, Repli-seq B-ALL in red and Repli-Capture-seq in dark red. (C) Hierarchical clustering analysis of all patient data sets based on their genome-wide RT programs confirms that there is no bias from the different methods exploited for RT profiling.
Methods for genome-wide RT profiling. (A) Bone marrow samples were collected from pediatric BCP-ALL patients and analyzed by 1 of the methods shown. Samples with high viability were analyzed by early/late (E/L) RT methods, and samples with poor viability were analyzed either by S/G1 methods or they were expanded in patient-derived xenografts (PDXs) to rejuvenate their viability for analysis by E/L RT methods.14 RT was measured using either E/L or S/G1 methods.14,28 In the E/L method, asynchronously growing cells are pulse-labeled with 5′-bromo-2′-deoxyuridine (BrdU) and separated by FACS into early and late S phase populations. BrdU-substituted DNA is immunoprecipitated with an anti-BrdU antibody and subjected to either microarray analysis (E/L Repli-ChIP16 or NextGen sequencing [E/L Repli-seq17 ]) to obtain an RT ratio [= log2(early/late)] along each chromosome. For the S/G1 method, unlabeled cells are sorted into S and G1 phase fractions, the relative abundance of genomic sequences is quantified by either microarray (S/G1 Repli-ChIP) or evenly spaced segments of genomic DNA are captured using oligonucleotides and subject to NGS (Repli-capture-seq). (B) Exemplary RT profiles showing the similarity in results using the various methods after appropriate local polynomial smoothing (Loess). Repli-Chip normal B-cell sanples are shown in grey, Repli-Chip B-ALL in orange, Repli-seq normal B-cells in black, Repli-seq B-ALL in red and Repli-Capture-seq in dark red. (C) Hierarchical clustering analysis of all patient data sets based on their genome-wide RT programs confirms that there is no bias from the different methods exploited for RT profiling.
Comparison of RT programs from leukemia patient samples and cell lines with one another using the same pipeline revealed 3540 50-kb variable segments (6.3% of the autosomal genome) that showed significant differences and generated 14 RT signatures (Figure 3A). Six signatures were linked to patients and cell lines containing the t(1;19) translocation that encodes the TCF3-PBX1 fusion protein (1 to 6 in Figure 3A-B). TCF3 (also known as E2A) is a transcription factor required for normal B- (and T-) cell differentiation and has been implicated in many lymphoid malignancies.29,30 PBX1 is a proto-oncogene with a critical role in hematopoiesis and lymphopoiesis.31 The TCF3-PBX1 fusion gene is believed to be a driver mutation in the BCP-ALLs in which it is found.32 RT signatures 1 and 2 were early replicating and 6 was late replicating uniquely in the TCF3-PBX1–positive samples. The fact that all 3 TCF3-PBX1–positive cell lines shared the same RT signatures as the patients’ TCF3-PBX1–positive leukemic cells underscores the epigenetic stability of their identified RT signatures. RT signatures 3, 4, and 5 substratified the TCF3-PBX1–positive samples. RT signature 8 was early replicating in a group of BCP-ALL samples of unknown genotype. Interestingly, RT signatures 7 and 9 were shared between BCP-ALL and certain AML samples and contained features of early myeloid differentiation (supplemental Figure 3). RT signatures 10, 11, and 12 identified subsets of BCP-ALL samples. Signature 13 was late in 1 case and 14 was late in 2 others. The exclusivity of these RT signatures for specific subsets of patients can be further probed by clustering all patient samples using only the 50-kb chromosomal segments (features) found in each of the RT signatures. For example, clustering analysis of patient samples using the chromosomal segments from RT signature 1 identified the patients carrying the TCF3-PBX1 translocation (Figure 3C). Heat maps and dendrograms for each RT signature from Figure 3A are shown in supplemental Figure 4.
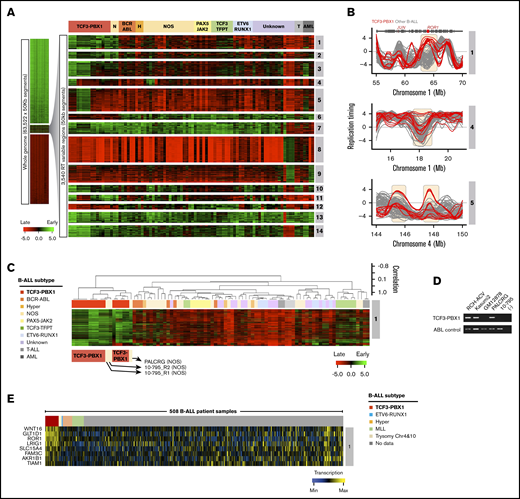
Establishment of BCP-ALL RT signatures. (A) Patient BCP-ALL stratification using RT profiling data. RT signatures are first identified by unsupervised k-means clustering of all 50-kb genomic segments that change between early and late replication from RT log2 ratio of +0.5 or more to/from –0.5 or less, respectively.7 These stringent criteria identify features that replicate significantly differently in specific clusters of patients (P < 2 × 10−16 using Student t tests). Branches of the dendrogram were constructed on the basis of correlation values (distance = 1 minus the correlation value). (B) Exemplary RT profiles of RT signatures. The top panel show an example of an RT feature (containing the ROR1 gene) of TCF3-PBX1–positive (red) vs TCF3-PBX1-negative (gray) BCP-ALL cells. (C) Hierarchical clustering of BCP-ALL samples for RT signature 1. (D) Reverse transcription polymerase chain reaction (RT-PCR) to determine the TCF3-PBX1 fusion messenger RNA (mRNA) in patient samples classified as NOS (PALCRG and 10-795). Specific primers for the TCF3-PBX1 fusion mRNA and for ABL as a control were used. Two TCF3-PBX1–positive cell lines were used as positive controls (RCH-ACV and Kasumi2). Normal B-lymphoid cells (GM12878) were used as a negative control. (E) Heatmap of expression patterns for genes within the RT signatures 1. Chr, chromosome; H, hyperdiploid; N, NOS; T, T-cell acute lymphoblastic leukemia.
Establishment of BCP-ALL RT signatures. (A) Patient BCP-ALL stratification using RT profiling data. RT signatures are first identified by unsupervised k-means clustering of all 50-kb genomic segments that change between early and late replication from RT log2 ratio of +0.5 or more to/from –0.5 or less, respectively.7 These stringent criteria identify features that replicate significantly differently in specific clusters of patients (P < 2 × 10−16 using Student t tests). Branches of the dendrogram were constructed on the basis of correlation values (distance = 1 minus the correlation value). (B) Exemplary RT profiles of RT signatures. The top panel show an example of an RT feature (containing the ROR1 gene) of TCF3-PBX1–positive (red) vs TCF3-PBX1-negative (gray) BCP-ALL cells. (C) Hierarchical clustering of BCP-ALL samples for RT signature 1. (D) Reverse transcription polymerase chain reaction (RT-PCR) to determine the TCF3-PBX1 fusion messenger RNA (mRNA) in patient samples classified as NOS (PALCRG and 10-795). Specific primers for the TCF3-PBX1 fusion mRNA and for ABL as a control were used. Two TCF3-PBX1–positive cell lines were used as positive controls (RCH-ACV and Kasumi2). Normal B-lymphoid cells (GM12878) were used as a negative control. (E) Heatmap of expression patterns for genes within the RT signatures 1. Chr, chromosome; H, hyperdiploid; N, NOS; T, T-cell acute lymphoblastic leukemia.
Interestingly, RT signatures 1, 2, and 6 (Figure 3A) identified 2 BCP-ALLs exhibiting an RT signature of TCF3-PBX1–positive cells despite the samples being designated as not otherwise specified (NOS; cannot be classified into typical genetic subtypes, including those with a t[1;19] karyotype). To determine whether these samples might have an undetected TCF3-PBX1 fusion, we performed reverse transcription polymerase chain reaction on messenger RNA isolated from these samples. Interestingly, this revealed a TCF3-PBX1 fusion messenger RNA in sample PALCRG but not in sample 10-795 (Figure 3D), which also did not contain a TCF3-TFPT fusion gene (supplemental Table 1).33 Next, we analyzed the gene expression patterns within these RT signatures using transcriptome data from 508 B-cell ALL (B-ALL) samples generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) program from patients enrolled in Children’s Oncology Group (COG) clinical trials.34 This analysis identified genes with coordinated changes in transcriptional activity and RT (Figure 3E; supplemental Figure 5). Among the genes overexpressed in RT signature 1, we identified WNT16, GLT1D1, ROR1, and others (Figure 3E). WNT16 has been shown to be a target of the TCF3-PBX1 fusion protein and is associated with the leukemogenesis of this BCP-ALL subtype.35 GLT1D is also upregulated in TCF3-PBX1–positive cells but not in patients with TCF3-HLF translocation.36 ROR1 encodes for the receptor orphan tyrosine kinase receptor 1, and it enhances the viability of TCF3-PBX1–positive cells.37,38 Supplemental Table 3 contains the complete gene list for all B-ALL RT signatures shown in Figure 3C.
To investigate the possible role of the TCF3-PBX1 fusion protein in determining the RT signatures of TCF3-PBX1–positive leukemic cells, we transfected 2 TCF3-PBX1–positive cell lines (Kasumi and RCH-ACV) with a pair of TCF3-PBX1 siRNA oligonucleotides.39 Despite evidence of significant reduction of the TCF3-PBX1 RNA in both lines (Figure 4A-B), very strong genome-wide correlations in RT (≥0.85) were observed after downregulation of TCF3-PBX1 (Figure 4C-D), and no significant changes were observed in their RT profiles (Figure 4E-F). However, TCF3-PBX1 knockdown also leads to apoptosis39 , precluding analysis of longer-term effects. In addition, we overexpressed a TCF3-PBX1 complementary DNA in the mature GM12878 B-cell line (Figure 4G) and detected no change in its original RT profile (Figure 4H-I). Together, these results argue against a direct or continuing action of the TCF3-PBX1 protein on the RT features unique to TCF3-PBX1–positive leukemic cells.
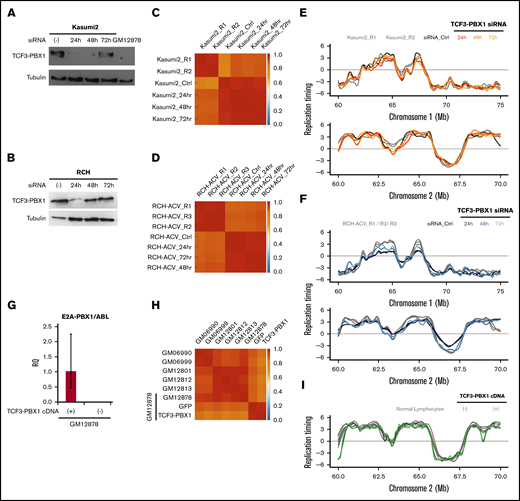
The TCF3-PBX1 fusion gene does not cause the RT alterations observed in TCF3-PBX1–positive cells. (A) Downregulation of the TCF3-PBX1 fusion protein with siRNA oligonucleotides in the Kasumi2 cell line. (B) Downregulation of TCF3-PBX1 fusion protein with siRNA oligonucleotides in the RCH cell line. (C-D) Genome-wide correlation matrices of Kasumi2 and RCH cells before and after downregulation of TCF3-PBX1 fusion protein. (E-F) Exemplary RT profiles at 2 distinct chromosomal regions confirms that the RT program is preserved after downregulation of TCF3-PBX1 fusion protein in Kasumi2 (E) and RCH (F) cell lines. (G) Overexpression of a TCF3-PBX1 complementary DNA (cDNA) in the normal B-cell line (GM12878). (H) Genome-wide correlation matrix of normal B cells with and without overexpression of the TCF3-PBX1 cDNA. (I) Exemplary RT profiles confirm that the RT program is preserved after overexpression of TCF3-PBX1 fusion protein in normal B cells. RQ, relative quantification.
The TCF3-PBX1 fusion gene does not cause the RT alterations observed in TCF3-PBX1–positive cells. (A) Downregulation of the TCF3-PBX1 fusion protein with siRNA oligonucleotides in the Kasumi2 cell line. (B) Downregulation of TCF3-PBX1 fusion protein with siRNA oligonucleotides in the RCH cell line. (C-D) Genome-wide correlation matrices of Kasumi2 and RCH cells before and after downregulation of TCF3-PBX1 fusion protein. (E-F) Exemplary RT profiles at 2 distinct chromosomal regions confirms that the RT program is preserved after downregulation of TCF3-PBX1 fusion protein in Kasumi2 (E) and RCH (F) cell lines. (G) Overexpression of a TCF3-PBX1 complementary DNA (cDNA) in the normal B-cell line (GM12878). (H) Genome-wide correlation matrix of normal B cells with and without overexpression of the TCF3-PBX1 cDNA. (I) Exemplary RT profiles confirm that the RT program is preserved after overexpression of TCF3-PBX1 fusion protein in normal B cells. RQ, relative quantification.
RT signatures of TCF3-PBX1–positive BCP-ALL cells suggest associations with central nervous system relapse
Although most TCF3-PBX1–positive BCP-ALL patients are cured with current therapies, ∼10% currently relapse with a high incidence of central nervous system (CNS) relapse.40-42 We analyzed WGS data (28× coverage) derived from 21 TCF3-PBX1–positive BCP-ALL patients at diagnosis by the St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. First, we identified differences in copy number variation and detected multiple amplified loci in all TCF3-PBX1–positive patients, but we did not detected differences that distinguished patients with CNS relapse (supplemental Figure 6). Next we exploited the deep coverage of this WGS data to generate RT profiles based on the fact that earlier-replicating sequences are higher in copy number than late-replicating sequences (Figure 5A), even without prior purification of S and G1 cells.20 RT data sets were derived from the 16 highest quality WGS data based on the correlation values between each other (supplemental Figure 6). RT profiles across all patient samples confirmed the high concordance of the RT programs derived from WGS data (Figure 5B). This group of TCF3-PBX1–positive patients included 2 patients who had an isolated CNS relapse and 1 who developed an unrelated secondary AML. Clustering analysis of variable chromosomal segments revealed 2 RT signatures shared by the 2 CNS relapse samples (Figure 5C) but not the nonrelapse or AML relapse samples (Figure 5C-D).
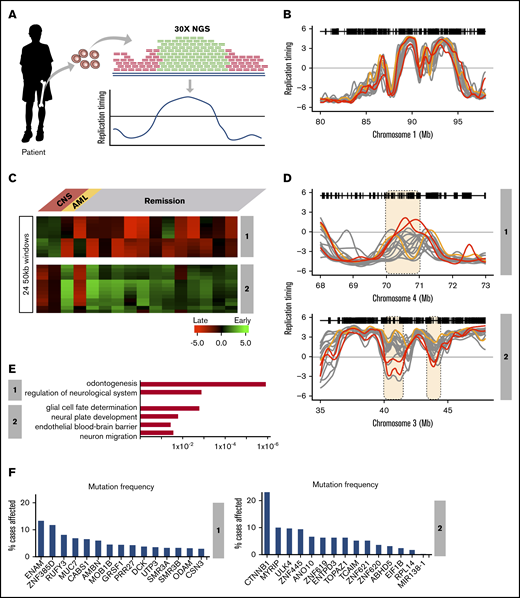
RT signatures linked to CNS relapse of TCF3-PBX1–positive BCP-ALL patients. (A) Patient samples were subjected to WGS at 28× depth. These sequences were subjected to a pipeline that converts their relative copy number into RT profiles.20 WGS data for 21 patients were obtained and the 16 with highest-quality RT profiles (based on genome-wide correlations to each other; supplemental Figure 4) were chosen for analysis. Of these, 2 samples were from patients who later suffered CNS relapse, and 1 was from a patient who developed a secondary AML; the remaining samples were from patients who were in remission. (B) Exemplary profiles of all patient samples from a typical genomic region that shows no variance between sample profiles. (C) Clustering analysis of differences between relapses vs remission. Two RT signatures distinguish patients who developed CNS relapse from those in remission. (D) RT profiles of each signature shown in panel C. (E) Ontology analysis of RT signatures shown in panel C. (F) Mutation frequencies for the genes in the RT signatures shown in panel C. Mutation frequencies were obtained from the National Cancer Institute Genomic Data Commons Data Portal.45
RT signatures linked to CNS relapse of TCF3-PBX1–positive BCP-ALL patients. (A) Patient samples were subjected to WGS at 28× depth. These sequences were subjected to a pipeline that converts their relative copy number into RT profiles.20 WGS data for 21 patients were obtained and the 16 with highest-quality RT profiles (based on genome-wide correlations to each other; supplemental Figure 4) were chosen for analysis. Of these, 2 samples were from patients who later suffered CNS relapse, and 1 was from a patient who developed a secondary AML; the remaining samples were from patients who were in remission. (B) Exemplary profiles of all patient samples from a typical genomic region that shows no variance between sample profiles. (C) Clustering analysis of differences between relapses vs remission. Two RT signatures distinguish patients who developed CNS relapse from those in remission. (D) RT profiles of each signature shown in panel C. (E) Ontology analysis of RT signatures shown in panel C. (F) Mutation frequencies for the genes in the RT signatures shown in panel C. Mutation frequencies were obtained from the National Cancer Institute Genomic Data Commons Data Portal.45
RT signature #1 contained features that were early replicating in the cells from the CNS relapse samples but late replicating in all other TCF3-PBX1–positive BCP-ALL patients. Gene ontology (GO) analysis of these chromosomal segments indicated that genes within this signature are associated with neurologic regulation (Figure 5E); specifically, this ontology term is linked to genes from the opiorphin family (SMR3A, SMR3B, and OPRPN), which are upregulated in head and neck squamous cell carcinoma.43 In addition, odontogenesis regulation was also associated with this RT signature, derived from genes AMBN, AMTN, ODAM, and ENAM, which are evolutionarily related genes all located within 500 kb of each other on chromosome 4.44 RT signature 2 contained features that were late replicating in samples from patients who developed CNS relapse but early replicating in samples derived from patients in remission. GO analysis indicated that genes within this signature were associated with glial cell fate determination (CTNNB1), neural plate development (CTNNB1), establishment of endothelial blood-brain barrier (CTNNB1), and regulation of neuron migration (ULK4) (Figure 5E). CTNNB1 and ULK4 are located within 500 kb of each other on chromosome 3 (Figure 5D). CTNNB1 encodes for β-catenin, is among the most frequently mutated genes in human cancer,45 and has the highest mutation frequency within RT signatures 1 and 2 (Figure 5F). Importantly, other genes reside in the domains with these GO term-driving genes. However, given the limited scope of this preliminary study, further studies will be required to establish the significance of a potential link between these genes and cellular phenotypes. Supplemental Table 4 contains the complete gene list of these RT signatures.
RT signatures of NOS BCP-ALL also suggest associations with clinical relapse
A total of 120 NOS patients’ samples were obtained from the COG bank. Sixty of these were from patients classified as high risk, 30 were from patients who relapsed within 3 years, and 30 were from patients who were still in remission after at least 3 years. Because most of these samples had too few cells to support the generation of high-quality RT profiles, we selected the 5 highest quality data sets each from paired sets of relapse and remission samples and generated RT signatures (Figure 6A). This analysis revealed 4 RT relapse-specific signatures, one of which was found in all relapses, one in 3 relapse samples, and 2 in 2 relapse samples (Figure 6B). For all 4 signatures, all remission samples were identical. Thus, despite the high degree of heterogeneity of the small number of NOS patients’ samples analyzed, their relapse RT signatures showed several interesting features. GO analysis identified genes in their RT signatures associated with interleukin-5 signaling and B-cell activation for RT signature 1; leukocyte tethering, interleukin-35, and interleukin-21 signaling for RT signature 3 (Figure 6C); and GO terms derived from IL5RA and ITPR1 genes, which are required for B-cell development and have the highest mutation frequency within the NOS RT signatures (Figure 6D). A complete list is provided in supplemental Table 5.
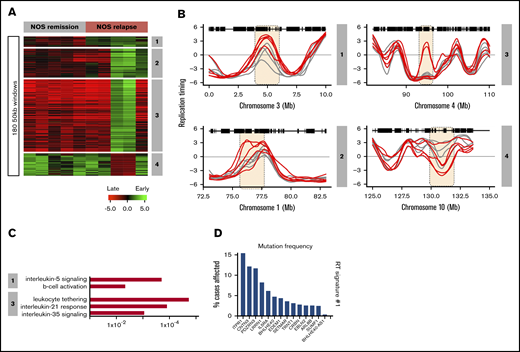
RT signatures of NOS patient relapse. Sixty NOS samples (30 relapse; 30 remission) were obtained from the Children’s Oncology Group (COG). Many of these samples were nonviable and had to be analyzed by the Repli-capture-seq S/G1 method (Figure 2), which frequently yielded a low signal-to-noise ratio. The 5 highest-quality relapse and remission samples (based on their correlation to normal B cells) were subjected to hierarchical clustering to identify RT signatures. (A) Four RT signatures distinguish patients with relapse from those in remission. Signature 1 includes all relapses, 2 includes only 3 relapses, and 3 and 4 are specific for 2 relapses. (B) RT profiles of each signature shown in panel A. (C) Ontology analysis of RT signatures shown in panel A. (D) Mutation frequencies for the genes in RT signature 1 shown in panel A. Mutation frequencies were obtained from the National Cancer Institute Genomic Data Commons Data Portal.45
RT signatures of NOS patient relapse. Sixty NOS samples (30 relapse; 30 remission) were obtained from the Children’s Oncology Group (COG). Many of these samples were nonviable and had to be analyzed by the Repli-capture-seq S/G1 method (Figure 2), which frequently yielded a low signal-to-noise ratio. The 5 highest-quality relapse and remission samples (based on their correlation to normal B cells) were subjected to hierarchical clustering to identify RT signatures. (A) Four RT signatures distinguish patients with relapse from those in remission. Signature 1 includes all relapses, 2 includes only 3 relapses, and 3 and 4 are specific for 2 relapses. (B) RT profiles of each signature shown in panel A. (C) Ontology analysis of RT signatures shown in panel A. (D) Mutation frequencies for the genes in RT signature 1 shown in panel A. Mutation frequencies were obtained from the National Cancer Institute Genomic Data Commons Data Portal.45
Leukemia-specific RT signatures match in part to RT profiles of normal human hematopoietic cell types
Comparison of BCP-ALL RT signatures with the RT changes during normal hematopoiesis revealed similarities with specific stages of B-cell development (supplemental Figure 3). To identify features of the leukemic cell RT signatures that might match those of normal hematopoietic cells and/or subsets of them at different stages of differentiation, we performed a hierarchical clustering analysis of the RT data for the subsets of genomic segments from the RT signature 1 that distinguish the TCF3-PBX1–positive samples, including the normal blood cell types (Figure 3A). The genomic regions from this RT signature display heterogeneous patterns of RT in mature B cells and T cells (Figure 7A). Further division of this subset of genomic regions by k-means clustering analysis (Figure 7B) revealed 3 groups of features within signature 1, 1 shared with normal pre-B cells (late B-precursors) but not normal pro-B or mature B cell lines (1A in Figure 7A-B), a second set shared with mature B-cell lines but not normal pre- or pro-B cells (1B in Figure 7A-B), and a third set not shared with any of the nonleukemic B cells analyzed (1C in Figure 7A-B). When these 3 sets of features were then used to subdivide signature 1, the TCF3-PBX1–positive patients’ cells and cell lines and related patient samples continued to form a closely related cohort. In addition, TCF3-PBX1–positive patient samples also clustered tightly with the RT features of the pre-B-cell samples using signature 1A and with the B-cell lines using signature 1B (Figure 7B). Importantly, signature 1A includes the gene for ROR1 (Figure 3B; supplemental Table 2). ROR1 replicates early in TCF3-PBX1–positive patients’ cells but late in all other BCP-ALL subtypes as well as in normal B cells (Figure 3C). ROR1 is highly expressed in t(1;19) BCP-ALL blasts and is required for their viability, even though ROR1 is not directly regulated by either the TCF3-PBX1 fusion protein or the pre-B-cell receptor.37,38 Moreover, ROR1 switches transiently to early replication in late B-cell progenitors (CD19+CD34–) before changing back to late replication in mature B cells during normal B-cell differentiation (Figure 3C), suggesting that TCF3-PBX1–positive patients’ cells retain the early RT program of normal pre-B cells (late B-precursors).
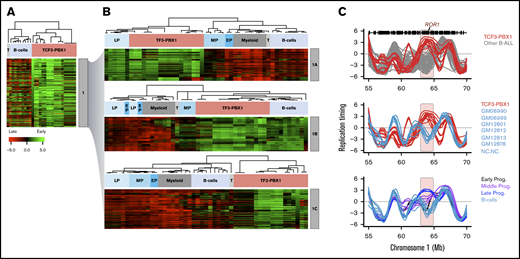
Analysis of RT abnormalities of TCF3-PBX1–positive cells. (A) Hierarchical analysis of RT signature 1 from Figure 3 that clusters TCF3-PBX1–positive cells with normal B cells. This RT signature can be subclassified in 3 RT subsignatures. (B) RT signature 1A: TCF3-PBX1–specific RT changes that track with normal pre-B cells (late B-precursors; CD34–CD19+ROR1+ cells). RT signature 1B: TCF3-PBX1–specific RT changes that track with Epstein-Barr virus immortalizedB cells. RT signature 1C: TCF3-PBX1–specific RT changes that do not track with any of the B cells and serve to substratify patient samples. (C) TCF3-PBX1–positive patient cells retain altered early replication of ROR1 from normal late B-precursors.
Analysis of RT abnormalities of TCF3-PBX1–positive cells. (A) Hierarchical analysis of RT signature 1 from Figure 3 that clusters TCF3-PBX1–positive cells with normal B cells. This RT signature can be subclassified in 3 RT subsignatures. (B) RT signature 1A: TCF3-PBX1–specific RT changes that track with normal pre-B cells (late B-precursors; CD34–CD19+ROR1+ cells). RT signature 1B: TCF3-PBX1–specific RT changes that track with Epstein-Barr virus immortalizedB cells. RT signature 1C: TCF3-PBX1–specific RT changes that do not track with any of the B cells and serve to substratify patient samples. (C) TCF3-PBX1–positive patient cells retain altered early replication of ROR1 from normal late B-precursors.
Discussion
Our results demonstrate the potential of RT profiles to identify markers characteristic of BCP-ALL subtypes and to substratify patient samples by outcome. This included the identification of several groups of domains that replicate together uniquely and consistently in TCF3-PBX1–positive leukemic cells compared with other BCP-ALL subsets (RT signatures). Evidence that these reflect a retained epigenetic state of the normal cells they resemble is provided by the demonstration of a subset of individual domains (RT features) with correlates in normal CD19+CD34– pre-B cells, but not more primitive normal CD19+CD34+ pro-B cells or more mature B cells. Thus, additional RT analysis of lymphoid progenitors present in fetal liver or adult sources would be of interest to extend these comparisons to other developmental stages of human hematopoiesis. Correlates shown here for CB-derived cells included ROR1, a gene that is uniquely expressed in the TCF3-PBX1–positive subtype of BCP-ALL where it is a therapeutic target.37 We also discovered signatures that substratify TCF3-PBX1–positive patient samples and RT signatures specific to and shared by TCF3-PBX1–positive as well as NOS BCP-ALL relapse cells. Other subtypes of BCP-ALL cells also contain distinguishing features. We conclude that RT profiles of BCP-ALL cells have stable prevalent clonotypic features operative in the normal phenotypes they resemble as well as others with potential prognostic import.
We also report several methodologies for analyzing and comparing normalized patterns of RT in normal differentiating human hematopoietic cells and leukemic samples from BCP-ALL patients. When possible, the method of choice is E/L Repli-seq,17 which yields consistently high-quality data from small numbers of cells. Samples with low viability can either be expanded as PDXs to regenerate their viability14 or RT profiles can be generated by a copy number comparison of reads in S vs G1 phase cells. Finally, when sufficiently deep WGS is available, RT profiles can be derived from total read copy number.20
RT often correlates with transcription but can clearly detect abnormalities not revealed by transcriptome analysis.11 The recent identification of cis-elements of RT control known as early replication control elements reveals that RT, 3D architecture, and subnuclear positioning are coregulated.46 Moreover, RT changes seen in leukemia reflect changes seen during normal development13 that are coordinated with changes in genome architecture.47 Thus, RT signatures that reflect alterations in epigenetic regulation of large-scale genome structure and function and are relatively easy to generate, may offer a novel genre of prognostic biomarkers in human leukemia.
Acknowledgments
The authors thank R. Didier for expert help with flow cytometry, and M. Hale and G. Edin and the British Columbia Cancer Agency Stem Cell Assay Laboratory staff for technical assistance in CB cell processing and animal irradiations.
This work was supported by National Institutes of Health (NIH), National Cancer Institute (NCI) grant R21 CA161666 and NIH, National Institute of General Medical Sciences grant R01 GM083337, and a Margaret and Mary Pfeiffer Professorship for Cancer Research (D.M.G.), a Terry Fox Foundation New Frontiers Program Project grant, and Canadian Cancer Research Society Research Institute grants (C.J.E.). D.J.H.F.K. held a Canadian Institutes of Health Research (CIHR) Vanier Scholarship, and C.A.H. held a CIHR Frederick Banting and Charles Best Doctoral Scholarship. Children’s Oncology Group trials were supported by NIH, NCI grants U10 CA98543, U10 CA98413, U10 CA180886, 1U24-CA196173, and U10 CA180899, and by St. Baldrick’s Foundation.
Authorship
Contribution: J.C.R.-M., D.M.G., and C.J.E. designed the research; J.C.R.-M., T.S., C.T.-G., N.N., D.J.H.F.K., C.A.H., K.N.K., V.S., and D.M.G. performed experiments; B.H.C., J.W.T., M.D., B.J.D., T.A.G., and C.J.E. provided patient samples; J.C.R.-M., J.Z., M.D., and A.K. analyzed and interpreted the data; and J.C.R.-M. and D.M.G. wrote the manuscript with input from all authors.
Conflict-of-interest disclosure: B.J.D. served on the scientific advisor board for Aileron Therapeutics, ALLCRON, Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, and EnLiven Therapeutics; served on the scientific advisory board and owns stock in Aptose Biosciences, Blueprint Medicines, β Cat, and Third Coast Therapeutics; served on the Board of Directors and owns stock in Amgen; served on the Board of Directors of the Burroughs Wellcome Fund and CureOne; served on the Joint Steering Committee for Beat AML LLS; was a founder of VP Therapeutics; received clinical trial funding from Novartis, Bristol-Myers Squibb, and Pfizer; and received royalties from Patent 6958335 (Novartis exclusive license) and from Oregon Health & Science University and Dana-Farber Cancer Institute (1 Merck exclusive license). The remaining authors declare no competing financial interests.
Correspondence: David M. Gilbert, Department of Biological Science, Florida State University, 319 Stadium Dr, Tallahassee, FL 32306-4295; e-mail: gilbert@bio.fsu.edu.

