

Key Points
The COX inhibitor indomethacin delays progression of ALL in a human xenograft mouse model.
The xenograft-derived ALL cells treated with indomethacin express elevated levels of p53.

Abstract
Acute lymphoblastic leukemia (ALL) develops in the bone marrow in the vicinity of stromal cells known to promote tumor development and treatment resistance. We previously showed that the cyclooxygenase (COX) inhibitor indomethacin prevents the ability of stromal cells to diminish p53-mediated killing of cocultured ALL cells in vitro, possibly by blocking the production of prostaglandin E2 (PGE2). Here, we propose that PGE2 released by bone marrow stromal cells might be a target for improved treatment of pediatric ALL. We used a xenograft model of human primary ALL cells in nonobese diabetic-scid IL2rγnull mice to show that indomethacin delivered in the drinking water delayed the progression of ALL in vivo. The progression was monitored by noninvasive in vivo imaging of the engrafted leukemic cells, as well as by analyses of CD19+CD10+ leukemic blasts present in spleen or bone marrow at the termination of the experiments. The indomethacin treatment increased the level of p53 in the leukemic cells, implying that COX inhibition might reduce progression of ALL by attenuating protective paracrine PGE2 signaling from bone marrow stroma to leukemic cells.
Introduction
B-cell precursor acute lymphoblastic leukemia (ALL) is the most prevalent pediatric cancer.1 Because of improved multimodal chemotherapy regimens over the past decades, the general survival of pediatric ALL is now close to 90%. There are, however, subgroups with poor prognosis, and ALL is still 1 of the most common causes of cancer-related deaths in children.2 Furthermore, pediatric ALL survivors often suffer from severe long-term side effects of the harsh chemotherapy treatments.3 For these reasons, there is a steady search for new strategies to improve the treatment of this disease.
The gene product of TP53 is regarded as an important barrier against cancer development,4 as well as being imperative for successful cancer treatment.5,6 The tumor suppressor p53 is a key factor in DNA damage responses, and it exerts most of its effect as a transcription factor.7 Accordingly, it is presumed that progression of cancer involves restraining the function of p53, either by mutations resulting in reduced stability of the protein, or by inactivating mutations.8 Although the majority of pediatric cases of ALLs express wildtype p53 at the time of diagnosis,9-11 gain-of-function mutations in the E3 ligase HDM2 that promotes degradation of p53 are frequent.12 Still, it is reasonable to assume that progression of ALLs might also involve additional ways of mitigating the levels and functions of wild-type TP53.
We have previously shown that activation of the cyclic adenosine monophosphate (cAMP)/PKA pathway in ALL cells promotes degradation of p53 at the protein level via enforced interaction between p53 and HDM2, resulting in reduced DNA damage–mediated killing of the cells.13-15 ALL is derived from various stages of immature B-cell precursors in the bone marrow,16 and the disease develops in close contact with stromal cells in bone marrow niches.17,18 Increasing evidence suggests that bone marrow stromal cells are recruited to tumor sites where they can contribute to tumor development, immunomodulation, and to treatment resistance.19,20 We demonstrated that stromal cells, when cocultured with patient-derived ALL cells, mimic the effects of cAMP/PKA-stimulating factors such as forskolin and 8-CPT-cAMP. Thus, both DNA damage-induced p53 and apoptosis were reduced in the leukemic cells cocultured with bone marrow stromal cells. Interestingly, the cyclooxygenase (COX) inhibitor indomethacin added to the cocultures attenuated the protective effect of the stromal cells. COX is 1 of the enzymes in the cascade responsible for converting arachidonic acid to eicosanoids such as prostaglandins.21 We22 and others23 have shown that bone marrow stromal cells produce prostaglandin E2 (PGE2), suggesting that the protecting effects of the bone marrow stroma on DNA damage-induced killing of ALL cells may involve PGE2.
Here we present COX as a putative target for improved treatment of ALL by demonstrating the ability of indomethacin to delay the progression of ALL in a xenograft model of primary patient-derived ALL cells in nonobese diabetic-scid IL2rγnull (NSG) mice.
Materials and methods
Cell culture and primary cell isolation
The ALL cell line REH24 was cultured as previously described.25 REH cells were originally derived from a 15-year-old female, and the cytogenetics of the cells is t12;21.24 HEK293T26 cells were cultured in Dulbecco’s modified Eagle medium/F-12-Glutamax (Thermo Fisher, Waltham, MA) supplemented with 10% fetal bovine serum (FBS), 125 U/mL penicillin, and 125 µg/mL streptomycin. The HEK293T cells were subcultured every 2 to 3 days upon reaching 70% to 90% confluence. Primary ALL blasts from newly diagnosed children were isolated as previously described,13 and the number of cells isolated from each patient varied substantially. The samples included in this study were selected based on whether sufficient numbers of cells were available for establishing the xenografts. Characteristics of the ALL cells from each patient included in this study are presented in Table 1. The collection of bone marrow aspirates was performed after informed consent by parents, in accordance with the Declaration of Helsinki. The collection of material was approved by the Regional Ethics Committee of Norway region Sør-Øst C (REK 2014/883).
Lentivirus production in HEK293T cells
Lentiviral vectors containing genes coding for firefly luciferase and enhanced green fluorescent protein (EGFP) were produced by transfecting HEK293T with 8.3 µg of each of the plasmids: pMD2.G envelope plasmid, pCMVΔ8.91 packaging plasmid, and pSLIEW transfer plasmid.27 Cells were cultured to 70% to 90% confluence at the day of transfection. The cell culture media was changed approximately 1 hour before transfection. Transfection mixtures were prepared using the Calcium Phosphate Transfection Kit (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. Cell culture medium was changed 4 hours after transfection. Viral supernatants were collected after 2 days and concentrated using LentiX Concentrator (Takara Bio Inc., Kusatsu, Japan) at 4°C overnight, before the viruses were collected by centrifugation at 1500g for 45 minutes at 4°C. Pellets were suspended in a small volume (<1 mL) of cell culture medium and stored at −80°C. Frozen lentiviral stocks were titrated using REH cells by using the standard transduction protocol (see the following section).
Lentiviral transduction of ALL cells
Cells (5 × 105 cells per well) were seeded into 48-well plates with 4 mg/mL polybrene (Merck Millipore, Burlington, MA) present in the cell culture media. REH cells were seeded in RPMI1640 medium (Lonza, Basel, Switzerland) supplemented with 10% FBS and 125 U/mL penicillin and 125 µg/mL streptomycin, whereas patient-derived ALL cells were seeded in SFEM medium (Stem Cell Technologies, Vancouver, Canada) supplemented with 10% FBS, 20 ng/mL interleukin-3, and 10 ng/mL interleukin-7 (R&D Systems, Minneapolis, MN). Lentiviral concentrates were added to the cells, and spinfection was performed by centrifugating the plates at 900g for 50 minutes at 34°C. After spinfection, plates were transferred to a standard cell incubator (37°C, 5% CO2 in a humidified atmosphere) for 2 days before removing the viral particles by 2 repeated washes at 300g for 10 minutes at 4°C. A small aliquot was taken from these cells to analyze the amount of EGFP+ cells by flow cytometry. The remaining cells were subjected to intratibial (IT) injection into NSG mice.
Establishment of xenograft model
Transduced ALL cells (5 × 105) were injected into 6- to 8-week-old female NSG mice (The Jackson Laboratory, Bar Harbor, ME) anesthetized with isoflurane (induction; 4% to 5%, maintenance; 2% to 3%, oxygen flow; 300 mL/min). The proximal end of the tibia was exposed as the knee was kept in a flexed position. A 23G needle was used to drill a hole into the tibia before injecting the ALL cells (40 µL per animal) using a 31G insulin syringe. Mice were treated with general and local analgesia; 0.05 mg/kg Temgesic (Schlering-Plough, Kenilworth, NJ) and 1 to 2 mg/kg Marcain (AstraZeneca, Cambridge, United Kingdom) before IT injection. General analgesia treatments were repeated 6 to 8 hours after IT injection. Mice were treated with vehicle (0.05% dimethyl sulfoxide) or 7.5 µg/mL indomethacin (Sigma-Aldrich, St. Louis, MO) in the drinking water, with water changes every second day throughout the entire experiment, as indicated in the figures. Mice were housed under specific pathogenic-free conditions with food and water ad libitum. Health status was monitored daily, and all animal procedures were conducted according to the approval by the Norwegian Food Safety Authority under identification numbers 6626 and 16180.
In vivo imaging
Leukemic progression was monitored by noninvasive in vivo imaging using an IVIS Spectrum CT instrument (PerkinElmer, Waltham, MA). D-Luciferin (150 mg/kg, PerkinElmer) substrate was administered by intraperitoneal injection. After 9 minutes, 3 images were recorded with 1-minute intervals using the autoexposure setting. Mice were euthanized when showing heavy engraftment or symptoms above a predetermined humane end point.
Harvesting of splenocytes and bone marrow cells
At the end of experiments, spleens were harvested from the mice, pictures taken, and cells were isolated from the spleens by crushing them between 2 microscopic slides. Bone marrow was flushed from the femur and tibia of the mice. For estimation of percentage leukemic cells in the spleen or bone marrow samples, the cells were stained with antibodies against human CD19 (#348814, BD Biosciences, San Jose, CA) and CD10 (#312218, BioLegend, San Diego, CA).
Measurement of serum PGE2
At the end of experiments, blood was collected from mice by cardiac puncture, drained into regular microcentrifuge tubes, and incubated for 30 minutes at room temperature. After centrifugation at 14 000g for 5 minutes at 4°C, the supernatants containing serum were collected and stored at −80°C until further use. PGE2 was measured using the Prostaglandin E2 ELISA kit – Monoclonal (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s protocol.
Western immunoblot analysis of p53
Bone marrow cells or splenocytes collected from NSG mice were lysed in radioimmunoprecipitation assay buffer (150 mM NaCl, 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) for 20 minutes on ice, vortexing every 5 minutes. Supernatants were collected by centrifugation at 13 000g for 20 minutes at 4°C, and the protein concentration in the lysates was measured using the Pierce BCA Protein Assay Kit (ThermoFisher). Equal amounts of protein were loaded on a 4% to 20% Criterion TGX Precast Midi Protein Gel (Bio-Rad, Hercules, CA), and western immunoblot analysis was performed using Trans-Blot Turbo Midi PVDF Transfer Packs (Bio-Rad). Membranes were blocked in 5% dry milk for 1 hour before probing with the p53 DO-I antibody (1 µg/mL, #sc-126, Santa Cruz Biotechnology, Dallas, TX) or Calnexin (1:1000, #2433S, Cell Signaling Technology, Danvers, MA) at 4°C overnight. After 3 × 5 minutes washing in TBS with Tween 20, the membranes were incubated in secondary antibody goat anti-rabbit IgG-HRP (Bio-Rad #1706515) or goat anti-mouse IgG-HRP (Bio-Rad #1706516) for 45 minutes at room temperature, followed by 3 × 5 minutes washing in TBS with Tween 20. The membranes were exposed to the SuperSignal West Dura Extended Duration Substrate kit (ThermoFisher) according to the manufacturer’s protocol. Images were captured using a Syngene ChemiGenious camera and presented by the GeneSnap software tool (Syngene, Cambridge, United Kingdom). Intensity of protein bands was quantified using the GeneTool software (Syngene).
Statistical methods
All statistical analyses were performed using the GraphPad Prism 8 software (San Diego, CA). Tests for normality were performed using the Kolmogorov-Smirnov and Shapiro-Wilk tests. All datasets were found to be normally distributed, and the unpaired Student t test was therefore used to determine statistically significant differences between treatment groups. P < .05 was considered as statistically significant.
Results
Establishment of ALL xenografts in NSG mice
ALL-derived REH cells, or primary leukemic cells isolated from bone marrow aspirates of children diagnosed with ALL, were stably transfected with the firefly luciferase-EGFP vector by lentiviral transduction. The transfection efficiency varied, but was typically between 2% and 10% for primary patient-derived cells, whereas it was ∼90% for REH cells, as monitored by EGFP expression (Figure 1A). ALL cells were IT injected in NSG mice, and we followed the progression of ALL in the mice by noninvasive in vivo imaging of luminescence from the firefly luciferase expressed in the leukemic cells. As shown in Figure 1B, growth of leukemic cells from ALL#18 was notable 11 weeks after injecting the ALL cells, and the leukemia progressed for another 4 weeks until the animals were euthanized. It should be emphasized, however, that the kinetics of the leukemia progression varied between the different primary ALL donors (data not shown). All experiments were terminated when the animals displayed symptoms beyond a predetermined humane end point. After terminating the experiment presented in Figure 1B, cells were isolated from the bone marrow of the mice, and the percentage of CD19+CD10+ leukemic cells was monitored by flow cytometry analyses (Figure 1C, left). As shown in the right panel of Figure 1C, engraftment of ALL#18 increased the percentage of human CD19+CD10+ in the murine bone marrow by 91%, compared with bone marrow cells from healthy control mice. Furthermore, spleens were harvested and spleen weights recorded (Figure 1D, left). The right panel of Figure 1D shows that engraftment of ALL#18 increased the weight of the spleens by 81% compared with spleens from healthy control mice.
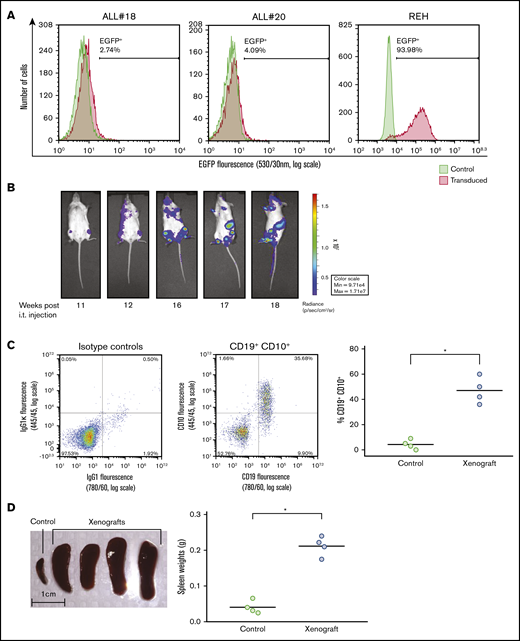
Establishment of xenograft model of ALL in NSG mice. (A) Lentiviral transduction efficiency of the pSLIEW vector into ALL#18, ALL#20, and REH cells was measured by flow cytometry analyses of EGFP fluorescence. (B-D) Four mice were IT injected with transduced ALL#18. (B) Progression of xenograft ALL#18 was followed by detection of luminescence intensity using noninvasive in vivo imaging, starting at 11 weeks after IT injection. (C) End point flow cytometry analysis of CD19+CD10+ leukemic cells in the bone marrow of xenograft ALL#18 mice. (Left) Dot plot presentation of CD19 vs CD10 expression of bone marrow cells from the mouse shown in panel B. (Right) The percentage of human CD19+CD10+ leukemic cells within the total bone marrow cell population of xenograft ALL#18 mice compared with noninjected (control) mice. Each circle represents the percentage of CD19+CD10+ cells in the bone marrow of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test). (D, left) Spleens from 4 xenograft ALL#18 mice and from 1 noninjected (control) mouse. (Right) Weight of spleens from the 4 xenograft mice compared with spleens from 4 control mice. Each circle represents the spleen weight (g) of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test).
Establishment of xenograft model of ALL in NSG mice. (A) Lentiviral transduction efficiency of the pSLIEW vector into ALL#18, ALL#20, and REH cells was measured by flow cytometry analyses of EGFP fluorescence. (B-D) Four mice were IT injected with transduced ALL#18. (B) Progression of xenograft ALL#18 was followed by detection of luminescence intensity using noninvasive in vivo imaging, starting at 11 weeks after IT injection. (C) End point flow cytometry analysis of CD19+CD10+ leukemic cells in the bone marrow of xenograft ALL#18 mice. (Left) Dot plot presentation of CD19 vs CD10 expression of bone marrow cells from the mouse shown in panel B. (Right) The percentage of human CD19+CD10+ leukemic cells within the total bone marrow cell population of xenograft ALL#18 mice compared with noninjected (control) mice. Each circle represents the percentage of CD19+CD10+ cells in the bone marrow of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test). (D, left) Spleens from 4 xenograft ALL#18 mice and from 1 noninjected (control) mouse. (Right) Weight of spleens from the 4 xenograft mice compared with spleens from 4 control mice. Each circle represents the spleen weight (g) of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test).
Indomethacin reduces the serum levels of PGE2 in NSG mice
Using in vitro models, we previously showed that ALL cells are exposed to PGE2 from bone marrow–derived stromal cells, and that PGE2 in turn lowers the levels of DNA damage-induced p53 and apoptosis in the ALL cells.22 Based on the ability of indomethacin to counteract the effects of stromal cells on cocultured ALL cells,22 we proposed that indomethacin might also counteract the effects of PGE2 in vivo. Before assessing the effects of indomethacin on ALL progression in NSG mice, we ensured that indomethacin indeed reduced the serum levels of PGE2 in vivo. Thus, indomethacin or vehicle was provided in the drinking water to NSG mice, and the levels of PGE2 were monitored after 10 weeks. As shown in Figure 2, indomethacin reduced the PGE2 levels in serum by 80%.
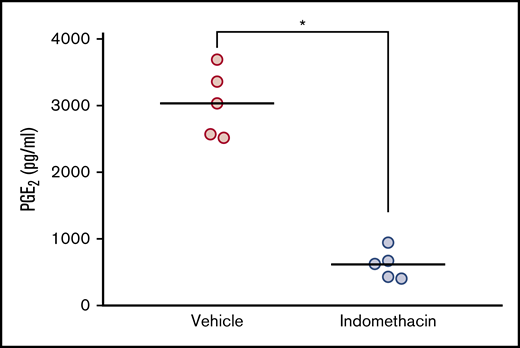
Indomethacin reduces the level of PGE2in mouse serum. Serum PGE2 measurement from serum of mice treated with and without indomethacin in their drinking water as described in “Materials and methods." Each circle represents the concentration of PGE2 (pg/mL) in the serum from 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test).
Indomethacin reduces the level of PGE2in mouse serum. Serum PGE2 measurement from serum of mice treated with and without indomethacin in their drinking water as described in “Materials and methods." Each circle represents the concentration of PGE2 (pg/mL) in the serum from 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test).
Indomethacin delays the progression of REH in vivo
REH cells transduced with the firefly luciferase-EGFP vector were injected into NSG mice. Indomethacin or vehicle was administered in the drinking water every second day throughout the entire experiment, and the growth of the cancer cells was followed by noninvasive in vivo imaging from 1 week after IT injection. As shown in Figure 3A-B, the luminescence from the leukemic cells treated with indomethacin was reduced by 81% 3 weeks after initiating the experiment. At 4 weeks after IT injection, the reduced growth of indomethacin-treated REH cells persisted, whereas the growth of REH cells in mice exposed to vehicle substantially increased.
![Indomethacin delays progression of REH in vivo. Xenograft REH mice (5 mice per treatment group) were treated with indomethacin or vehicle as described in “Materials and methods." (A) Progression of leukemia was followed by noninvasive in vivo imaging of luminescence from the mice. The images were taken between 1 and 4 weeks after IT injection as indicated. (B) Xenograft luciferase activity (photons per second [p/s]) from the xenografts presented in panel A measured relative to week 1 after IT injection. Data represent the mean ± standard error of the mean, n = 5, *P < .05 (unpaired Student t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/21/10.1182_bloodadvances.2019000473/5/m_advancesadv2019000473f3.png?Expires=1770038573&Signature=Ti2e~DAE9vnMNbijpRvBJ~mwvmTgrj0Y7Qsmm9BQxJFQ0jzMw10rnnOlNmTyJ1AlxncpcdAXG2pVgBkLIW98KzvyWbvFkLWV3kOCD6APd6~c7gKZ4j~lPM-gF7ZfEnxTMZcP1ivgfhdjg1tVQhad~mNhfHH8CQnCWjVDEYTwmUHxqpJGOjg6fm-9DwXLN5S-DLUUd3Lz3puX4jNCP9P~FXgtbh0nRkUAvlqwuLuh2VSgQbce5X9jVRqvwsTfdJHx3EnDtH1Tezl4UcEwNQ8z4D0oguH9pSFSEYi-kwX-6CaL0ma0y3LvW1Eo3O5LJ1O9rNc5Z3UPpGOI5xvQkjSKGA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Indomethacin delays progression of REH in vivo. Xenograft REH mice (5 mice per treatment group) were treated with indomethacin or vehicle as described in “Materials and methods." (A) Progression of leukemia was followed by noninvasive in vivo imaging of luminescence from the mice. The images were taken between 1 and 4 weeks after IT injection as indicated. (B) Xenograft luciferase activity (photons per second [p/s]) from the xenografts presented in panel A measured relative to week 1 after IT injection. Data represent the mean ± standard error of the mean, n = 5, *P < .05 (unpaired Student t test).
Indomethacin delays progression of REH in vivo. Xenograft REH mice (5 mice per treatment group) were treated with indomethacin or vehicle as described in “Materials and methods." (A) Progression of leukemia was followed by noninvasive in vivo imaging of luminescence from the mice. The images were taken between 1 and 4 weeks after IT injection as indicated. (B) Xenograft luciferase activity (photons per second [p/s]) from the xenografts presented in panel A measured relative to week 1 after IT injection. Data represent the mean ± standard error of the mean, n = 5, *P < .05 (unpaired Student t test).
Indomethacin delays the progression of primary ALL in vivo
Leukemic blasts from ALL#18 were transfected with the firefly luciferase-EGFP vector, and the ALL cells were injected into NSG mice. Indomethacin or vehicle was administered in the drinking water, and the progression of the leukemia was followed by noninvasive in vivo imaging of the mice. The images taken at the end of the experiment at day 72 (Figure 4A) indicated that indomethacin treatment of the mice reduced the progression of ALL#18. The mice were euthanized, and the percentage of leukemic cells in various organs was assessed by flow cytometry. Indomethacin was found to reduce the load of CD19+CD10+ cells in the bone marrow (Figure 4B, left) and spleen (Figure 4B, right) by 31% and 37%, respectively. The weights of the spleens were also reduced by indomethacin (Figure 4C), further supporting the inhibitory effect of COX inhibition on progression of ALL#18 in vivo. Finally, we collected spleens from xenografts of ALL#20 and #32 treated with indomethacin or vehicle, and again indomethacin reduced the load of leukemic blasts by 42% (ALL#20, Figure 4D) and 34% (ALL#32, Figure 4E). In supplemental Table 1, we have summarized the data for all the xenografts included in the study.
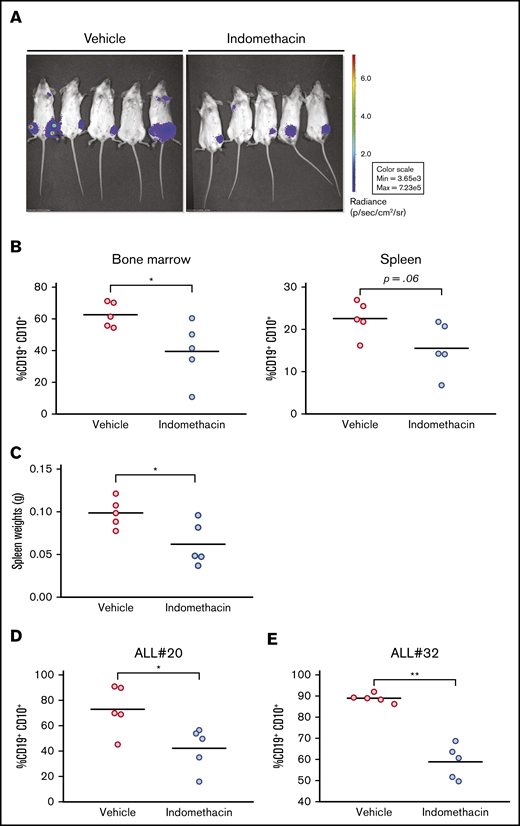
Indomethacin delays progression of ALL in vivo. (A) Xenograft ALL#18 mice (5 mice per treatment group) were treated with indomethacin or vehicle as described in “Materials and methods." Progression of leukemia was followed by noninvasive in vivo imaging of luminescence from the mice. The images were taken 10 weeks after IT injection. (B) End point flow cytometry analysis of human CD19+CD10+ leukemic cells in the total bone marrow (left) and spleen (right) cell populations of the mice presented in panel A. Each circle represents the percentage of human CD19+CD10+ cells in the total spleen or bone marrow cell populations of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .05 (unpaired Student t test). (C) Weight of spleens from the mice presented in panel A. Each circle represents the spleen weight (g) of 1 mouse. Horizontal lines represent the mean of each treatment group, *P = .05 (unpaired Student t test). (D-E) End point flow cytometry analysis of CD19+CD10+ leukemic cells in the bone marrow of xenograft ALL#20 (D) and ALL#32 (E) mice. Each circle represents the percentage of human CD19+CD10+ cells in the total bone marrow cell population of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .05, **P < .01 (unpaired Student t test).
Indomethacin delays progression of ALL in vivo. (A) Xenograft ALL#18 mice (5 mice per treatment group) were treated with indomethacin or vehicle as described in “Materials and methods." Progression of leukemia was followed by noninvasive in vivo imaging of luminescence from the mice. The images were taken 10 weeks after IT injection. (B) End point flow cytometry analysis of human CD19+CD10+ leukemic cells in the total bone marrow (left) and spleen (right) cell populations of the mice presented in panel A. Each circle represents the percentage of human CD19+CD10+ cells in the total spleen or bone marrow cell populations of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .05 (unpaired Student t test). (C) Weight of spleens from the mice presented in panel A. Each circle represents the spleen weight (g) of 1 mouse. Horizontal lines represent the mean of each treatment group, *P = .05 (unpaired Student t test). (D-E) End point flow cytometry analysis of CD19+CD10+ leukemic cells in the bone marrow of xenograft ALL#20 (D) and ALL#32 (E) mice. Each circle represents the percentage of human CD19+CD10+ cells in the total bone marrow cell population of 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .05, **P < .01 (unpaired Student t test).
Indomethacin enhances the levels of p53 in cells from bone marrow of xenograft mice
We previously proposed that PGE2 reduces DNA damage-induced apoptosis in cultured ALL cells by diminishing the p53 levels.22 To test our hypothesis that indomethacin exerts its inhibiting effect on leukemia progression by counteracting the stroma cell–mediated downregulation of p53, we performed western immunoblot analyses of p53 levels in cells collected from bone marrow samples of mice treated with or without indomethacin. We used antibodies specific for human p53 and the human control protein calnexin, ensuring that only p53 levels in the leukemic cells were monitored. Irradiated REH cells were included as a positive control for human p53 in the immunoblot presented in Figure 5 (left). As shown in the right panel of Figure 5A, indomethacin significantly enhanced the levels of p53 in the bone marrow cells of xenograft REH mice by 27%. Similar analyses of p53 expression in bone marrow cells from xenograft ALL#18 and xenograft ALL#20 mice revealed the p53 expression was enhanced by 46% (Figure 5B) and 71% (Figure 5C), respectively.
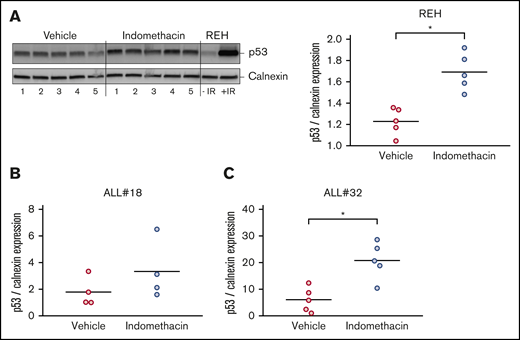
Indomethacin enhances p53 levels in bone marrow cells from xenograft mice. Bone marrow cells were harvested from xenograft REH mice, xenograft ALL#18 mice, or xenograft ALL#32 mice treated with indomethacin or vehicle for 4, 16, or 23 weeks, respectively. Cells were subjected to western immunoblot analysis with antibodies specific for human p53 as described in “Materials and methods." Calnexin was used as loading control. (A, left) Western immunoblot of p53 and calnexin expression from xenograft REH mice. Regular REH cells treated with and without irradiation (IR) were used as controls for human p53. (Right) Quantification from the immunoblot in the left panel of p53 band intensities relative to calnexin. Quantifications from immunoblots of p53 band intensities relative to calnexin from xenograft ALL#18 (B) and ALL#32 (C) mice. (A-C) Each circle represents the intensity of p53 relative to calnexin in 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test).
Indomethacin enhances p53 levels in bone marrow cells from xenograft mice. Bone marrow cells were harvested from xenograft REH mice, xenograft ALL#18 mice, or xenograft ALL#32 mice treated with indomethacin or vehicle for 4, 16, or 23 weeks, respectively. Cells were subjected to western immunoblot analysis with antibodies specific for human p53 as described in “Materials and methods." Calnexin was used as loading control. (A, left) Western immunoblot of p53 and calnexin expression from xenograft REH mice. Regular REH cells treated with and without irradiation (IR) were used as controls for human p53. (Right) Quantification from the immunoblot in the left panel of p53 band intensities relative to calnexin. Quantifications from immunoblots of p53 band intensities relative to calnexin from xenograft ALL#18 (B) and ALL#32 (C) mice. (A-C) Each circle represents the intensity of p53 relative to calnexin in 1 mouse. Horizontal lines represent the mean of each treatment group, *P < .01 (unpaired Student t test).
Discussion
In the present study, we have used a xenograft model of ALL in NSG mice to show that the COX inhibitor indomethacin delays the progression of ALL in vivo. By injecting primary ALL blasts or the ALL-derived cell line, REH, directly into the bone marrow of the mice, the engrafted human leukemic cells were exposed to factors produced by resident stromal cells. The xenograft model allowed us to follow the progression of ALL in vivo, and to monitor the impact of targeting bone marrow–derived components.
Our previous in vitro studies revealed that elevation of cAMP levels in ALL cells increases the survival of the cells by reducing p53 accumulation. This was achieved either by exposing the ALL cells to cAMP elevating compounds such as forskolin and 8-CPT-cAMP13-15 or by coculturing the ALL cells with bone marrow–derived stromal cells.22 We22 and others23 have shown that bone marrow stromal cells produce PGE2, which upon binding to its receptors, is able to elicit a cAMP response in the target cells.28,29 ALL cells are known to express the EP2 type of PGE2 receptors,19,20 and we showed that indomethacin countered the protective effects of stromal cells on the cocultured leukemic blasts. Thus, we proposed that the enhanced survival of leukemic cells was provided by PGE2 produced by the bone marrow stromal cells.22
Indomethacin is a nonsteroidal anti-inflammatory drug (NSAID) used for more than half a decade to treat symptoms of inflammation such as fever, pain, and stiffness.30 Based on our previous in vitro experiments,13-15,22 we proposed that indomethacin might be used in treatment of ALL. To test our hypothesis in vivo, we exposed the ALL xenograft mice to daily doses of indomethacin that were lower than the anti-inflammatory dosages commonly used in mice studies.31-33 As expected, we demonstrated that indomethacin reduced the serum levels of PGE2 in the xenograft mice by ∼80%. Furthermore, indomethacin significantly delayed the growth of the ALL xenografts of both primary leukemic blasts and the ALL-derived cell line REH. It is believed that early oncogenic events provide premalignant cells with slightly elevated levels of p53 as a guardian against further DNA damage-induced cancer development.34 Our previous finding that bone marrow–derived stromal cells reduce the levels of both basal and DNA damage-induced p53 in ALL cells22 implied that that bone marrow stromal cells might contribute to tumor progression by attenuating the p53 levels in the leukemic blasts. In support of this perception, we show here that indomethacin enhanced the levels of p53 in the leukemic cells isolated from the bone marrow of xenograft mice. We therefore propose that indomethacin delays the progression of ALL in the present xenograft model by countering the ability of bone marrow stroma to reduce the levels of p53.
The current favorable prognosis for children with ALL is based on successful development of multimodal chemotherapy.35 The backside of the coin is that these children often suffer from long-term side effects of the harsh treatment.36 Enhancement of the therapeutic index of current chemotherapeutic drugs is therefore highly desired. This would potentially allow lowering the effective doses of the drugs, and thereby reduce the devastating side effects of the treatments. Considering the critical role of p53 in DNA damage-based chemotherapy,37,38 we propose here that indomethacin may be useful as an adjuvant to increase the therapeutic index of DNA-damaging drugs. This notion is supported by our previous results showing that bone marrow stromal cells reduce DNA damage-induced p53 and apoptosis of ALL cells, and that indomethacin enhances DNA damage-induced apoptosis of leukemic cells cocultured with bone marrow stroma cells.22 However, although indomethacin is a widely used NSAID, its side effects after long-term use should not be neglected.30 One might therefore also consider alternative approaches to target the cAMP signaling pathway in leukemic cells. This could include protein kinase A inhibitors such as Rp-8-Br-cAMPs, or specific inhibitors of the functional EP2 (PGE2) receptors known to be expressed on ALL cells.19,20 Interestingly, it was previously shown that inhibiting adenylyl cyclase by the opioid dl-methadone enhanced the sensitivity of ALL cells to doxorubicin.39
NSAIDs have been proposed to reduce the risk of developing solid cancers.40,41 Although the antitumor effects of indomethacin colorectal cancers are documented,42,43 the mechanisms of action are not established. The role of indomethacin as an unselective COX inhibitor is clear, but COX-independent effects of indomethacin have also been documented, such as activation of the peroxisome proliferator-activated receptors γ and δ.44,45 It should also be noted that PGE2 might exert its known antiapoptotic effects via mechanisms other than enhancing cAMP levels, including acting via the EGFR,46,47 phosphatidylinositol 3-kinase/AKT,48 and extracellular signal-regulated kinase49 pathways. We therefore cannot exclude the possibility that indomethacin prevents the growth of ALL xenograft by mechanisms other than via targeting PGE2-induced cAMP signaling. Nevertheless, the novel findings of the present study remain: that a commonly used NSAID such as indomethacin can enhance the level of p53 in leukemic cells and delay the progression of ALL in vivo. Our results imply that targeting the cAMP signaling pathway might be useful to increase the therapeutic index of DNA-damaging drugs, and thereby potentially reduce the long-term side effects of current antileukemia treatment regimens.
Acknowledgments
The authors acknowledge the help of Nina-Cathrin Robinson in obtaining the bone marrow aspirates of newly diagnosed ALL patients. The authors thank the National Core Facility for Human Pluripotent Stem Cells, Norwegian Center for Stem Cell Research, Oslo University Hospital, for providing the necessary facilities to perform viral experiments. The authors also thank Olaf Heidenreich (Newcastle University) for the kind gift of the pSLIEW vector.
This work was supported by the University of Oslo, the Norwegian Cancer Society (PR-2006-0256), the UNIFOR FRIMED research fund, The Blix Family Legacy, the Anders Jahre Research foundation, and the Nansen Legacy. These organizations are public or nonprofit organizations that support science in general; they had no role in gathering, analyzing, or interpreting the data.
Authorship
Contribution: S.S., H.K.B., and A.F. designed the experiments; N.R., E.D., S.B., K.M.G., E.H.N., and S.S. performed experiments; N.R., E.D., S.S., S.B., and H.K.B. analyzed data; N.R. produced the figures; S.S. and H.K.B. supervised the project; E.R. and M.M.B. provided materials and recruited patients to the study; E.H.N., E.R., and M.M.B. provided important clinical insight; N.R. and H.K.B. wrote the original draft; and N.R., E.D., A.F., S.B., K.M.G., E.H.N., M.M.B., E.R., H.K.B., and S.S. reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Heidi Kiil Blomhoff, Department of Molecular Medicine, Institute of Basic Medical Sciences, University of Oslo, PO Box 1112 Blindern, N-0317 Oslo, Norway; e-mail: h.k.blomhoff@medisin.uio.no.

