

Key Points
Reexpansion of CAR T cells led to further investigations which confirmed the clonal nature of this expansion.

Introduction
Chimeric antigen receptor T-cell (CAR T-cell) therapy targeting CD19 is highly effective in B-cell acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma, with many patients achieving a durable remission.1-5 Following CAR T-cell infusion, peak expansion occurs over a period of several days, generally following an inflammatory phase, otherwise known as cytokine release syndrome (CRS).6 Following this peak expansion, there is typically a contraction in the number of CAR T cells, potentially followed by low-level persistence.1,7 Reexpansion of CAR T cells from the original infusion following the contraction phase, to a level exceeding the initial peak, is highly unusual, as is a clonal expansion.8 We report on a delayed clonal expansion in a patient receiving lentiviral vector-mediated, anti-CD22 CAR T-cell therapy to treat ALL. This clonal expansion occurred in the setting of residual 18 fluorodeoxyglucose (FDG)-avid extramedullary disease, with subsequent complete remission, which was followed by termination of this expansion.
Case description
A 28-year-old female with post–allogeneic hematopoietic stem cell transplantation and post-blinatumomab relapse of pre–B-cell ALL was enrolled on a phase 1 anti-CD22 CAR T-cell protocol. Pre-CAR evaluation revealed extensive CD22+ ALL involvement, with >90% replacement of the bone marrow with leukemic blasts, the presence of circulating peripheral blasts, and extensive FDG-avid extramedullary disease (including right cervical nodes, right parotid gland, right axillary nodes, pancreatic involvement, right omental masses, and right retro-orbital involvement [Figure 1A]). After lymphodepletion with fludarabine and cyclophosphamide from days −4 to −2, she received 1 × 106 transduced CAR T cells/kg on day 0. She developed fevers within 9 hours of the CAR infusion, consistent with infusion-related fevers, with onset of more persistent fevers and CRS starting on day +3. Tocilizumab (8 mg/kg) was given on day +6 for grade 2 CRS6 followed by a single dose of methylprednisolone (1 mg/kg), which led to rapid improvement and resolution of CRS. On day +11, she was restarted on methylprednisolone at 1 mg/kg every 12 hours for development of CAR T-cell-related coagulopathy that was transiently discontinued but subsequently resumed for recrudescent fevers and was weaned off by day +35, with full resolution of all symptoms.
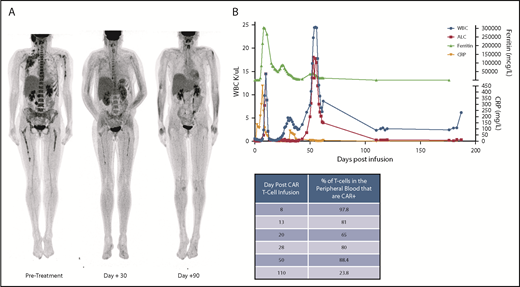
Clinical course. (A) FDG–positron emission tomography (PET) scan demonstrating significant extramedullary disease burden at pretreatment, followed by clinical response at day +30, and subsequent full resolution of any PET-avid disease at day +90. Subject was MRD− in the bone marrow by the day +30 evaluation and remained MRD− at the subsequent time points. (B) Clinical timeline with relevant laboratory findings. During the initial CRS, patient had a concurrent rise in CRP and ferritin which coincided with the initial WBC peak. During the second expansion phase, the patient remained clinically well without any fevers and had only modest increase in C-reactive protein (CRP) and ferritin, but with an even higher WBC than with the initial expansion. Concurrent table demonstrates the % of T cells which were CAR+ in the peripheral blood at the various timepoints.
Clinical course. (A) FDG–positron emission tomography (PET) scan demonstrating significant extramedullary disease burden at pretreatment, followed by clinical response at day +30, and subsequent full resolution of any PET-avid disease at day +90. Subject was MRD− in the bone marrow by the day +30 evaluation and remained MRD− at the subsequent time points. (B) Clinical timeline with relevant laboratory findings. During the initial CRS, patient had a concurrent rise in CRP and ferritin which coincided with the initial WBC peak. During the second expansion phase, the patient remained clinically well without any fevers and had only modest increase in C-reactive protein (CRP) and ferritin, but with an even higher WBC than with the initial expansion. Concurrent table demonstrates the % of T cells which were CAR+ in the peripheral blood at the various timepoints.
Her baseline white blood cell (WBC) count and absolute lymphocyte count (ALC) on day 0 before CAR T-cell infusion and following lymphodepleting chemotherapy were 590 K/µL and 130 K/µL, respectively (Figure 1B). By day +10, during CRS, her WBC count had increased to a peak of 14 480 K/µL containing entirely lymphocytes, with a neutrophil count of 0 K/µL. Corresponding flow cytometry during CRS identified 97.8% of the T cells were CD22 CAR+. Other notable features of her CAR T-cell expansion included swelling and pain at known extramedullary sites of disease, including unilateral right arm swelling and edema as well as right periorbital swelling.
On day +28, her WBC count and ALC were 2510 K/µL and 210 K/µL, respectively. Corresponding flow cytometry revealed a minimal residual disease (MRD)–negative complete remission in the blood and bone marrow, with 87% and 80% of T cells expressing the CD22 CAR, respectively. FDG positron emission tomography imaging demonstrated resolution of or decreasing FDG avidity of all areas of extramedullary disease.
At day +53, she was found to have an asymptomatic rapid rise in WBCs (from 7730 K/µL to a peak of 24 430 K/µL in a 4-day period) with a lymphocyte predominance (from an ALC of 5260 K/µL to 17 780 K/µL). Concurrent flow cytometry on day +50 demonstrated that 88.4% of the T cells were CD22 CAR+. Despite the absence of fevers, hypotension, or other signs consistent with clinical CRS, steroids were started at a dose of 0.5 mg/kg prednisone, given her recent complicated CRS course and rapid rise in WBCs. Steroids were weaned off over 5 days, followed by a marked decrease in WBC count. Concurrent Epstein-Barr virus and cytomegalovirus polymerase chain reaction (PCR) tests were negative. Molecular studies via T-cell receptor (TCR) sequencing at day +51 and +53 identified a dominant TCR clonotype (TCRBV20/TCRBD02-01*02/TCRBJ02-07*01 CSARDKASGRMYEQYF) (Figure 2A). Quantitative assessment of the TCR clonotype demonstrated the frequency of this clone was 16.7% of all circulating T cells on day +51, increasing to 44% at day +53.
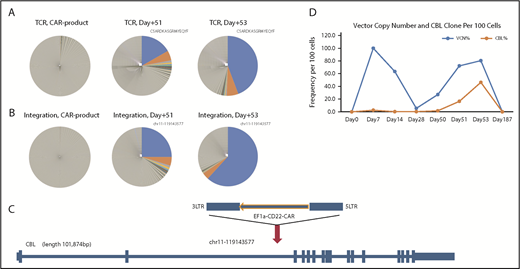
Expansion and persistence of CAR T cell with lentivector integrated in the CBL gene. (A) TCR diversity of CAR product before infusion; samples taken from the patient on days +51 and +53. A major clonotype that was obvious in day +51 and became more dominant on day +53. (B) Integration site diversity of CAR product before infusion and samples taken from the patient on days +51 and +53. The top clones at days +51 and +53 are the same and have same vector integrated in chr11 at position 119143577. (C) Details of the integration in the dominant clone at days +51 and +53. The lentiviral vector carrying the EF1a–anti-CD22 CAR integrated in the second intron of CBL gene on chr11, in the opposite orientation to the gene. (D) Longitudinal quantification of CAR vector copy numbers and the CBL clone in patient WBCs by ddPCR. The vector copy number (copies per 100 cells) shows 2 phases of expansion of CAR T cells. The first phase peaked at day +7 to +14 and subsided around day +30. The second phase peaked at day +53. The clone with the vector inserted into the CBL gene was quantified with ddPCR assay specific for the junction between the vector and host DNA. The clone is detectable, at a low level, from day +7 throughout day +53. During the second phase of CAR T expansion, the CBL clone became the dominant clone at day +53, constituting 46% of total WBCs in the patient and more than one-half of the CAR T cells.
Expansion and persistence of CAR T cell with lentivector integrated in the CBL gene. (A) TCR diversity of CAR product before infusion; samples taken from the patient on days +51 and +53. A major clonotype that was obvious in day +51 and became more dominant on day +53. (B) Integration site diversity of CAR product before infusion and samples taken from the patient on days +51 and +53. The top clones at days +51 and +53 are the same and have same vector integrated in chr11 at position 119143577. (C) Details of the integration in the dominant clone at days +51 and +53. The lentiviral vector carrying the EF1a–anti-CD22 CAR integrated in the second intron of CBL gene on chr11, in the opposite orientation to the gene. (D) Longitudinal quantification of CAR vector copy numbers and the CBL clone in patient WBCs by ddPCR. The vector copy number (copies per 100 cells) shows 2 phases of expansion of CAR T cells. The first phase peaked at day +7 to +14 and subsided around day +30. The second phase peaked at day +53. The clone with the vector inserted into the CBL gene was quantified with ddPCR assay specific for the junction between the vector and host DNA. The clone is detectable, at a low level, from day +7 throughout day +53. During the second phase of CAR T expansion, the CBL clone became the dominant clone at day +53, constituting 46% of total WBCs in the patient and more than one-half of the CAR T cells.
Disease restaging evaluations 2 months following this second expansion demonstrated ongoing MRD− remission in the blood and bone marrow with full resolution of FDG-avid extramedullary disease. Four months later (6 months postinfusion), the patient developed biopsy-proven CD22− extramedullary relapse at a periorbital site despite persistently detectable CD22+ CAR T cells by flow cytometry at a level of 4.8% of all T cells, suggesting that the mechanism of relapse was not due to CAR T-cell failure.
Because lentivectors randomly integrate in the host genome and the integration site can serve as a unique molecular barcode to track the clonal abundance, quantitative vector integration site analysis was performed to determine if the vector insertion site potentially contributed to the unexpected secondary clonal expansion of the CAR T cells. The results confirmed marked clonality at the second peak of CAR T-cell expansion, with the dominant T-cell clone on day +51 and day +53 containing a copy of the vector integrated in the second intron of CBL gene at chr11-119143577 (hg19) (Figure 2B-C), with the proviral insertion in the opposite orientation of the CBL gene. A Droplet Digital PCR (ddPCR) assay specifically designed for this CBL integration site junction was used to quantify the longitudinal frequency of this clone (Figure 2D). The clone containing the vector integrated in the CBL gene could be identified at day +7 post CAR T-cell infusion at a frequency of 2.6% of total WBCs and remained at a low frequency through the first 30 days. The frequency of this clone increased dramatically during the second CAR T expansion, with a peak frequency of 46.3% of total WBC count at day +53. By day +180 (6 months) following steroid treatment, the clone was undetectable, coinciding with the decline of total CAR T cells. No further reexpansion of the clone was seen.
To assess the extent of clonal expansion following CAR T infusion, we performed integration site analysis on samples from 7 additional patients enrolled on the anti-CD22 CAR T trial. We found no evidence of any clonal expansion of the vector-modified cells in the samples from these additional patients.
Methods
Clinical trial
This subject was enrolled on a phase 1 trial testing CD22 CAR T cells using a 3+3 dose-escalation design for the treatment of patients with relapsed/refractory CD22+ leukemia or lymphoma. The CD22 CAR T-cell construct and initial clinical trial results has been previously described.9-11 The subject provided written informed consent. All patients were treated in the Pediatric Oncology Branch of the National Cancer Institute, and the protocol was approved by the National Cancer Institute institutional review board and the National Institutes of Health Recombinant DNA Advisory Committee. The subject was also enrolled on an institutional review board-approved genomics protocol, which allowed for additional testing of blood samples. This trial was registered at clinicaltrials.gov as #NCT02315612.
TCR sequencing
ImmunoSEQ hsTCRB sequencing kit (Adaptive Biotechnologies, Seattle, WA) was used to profile TCR repertoire following the manufacturer’s protocol. Briefly, genomic DNA was amplified using multiplexed primers and sequenced on Illumina NextSeq. Data analysis of TCR clonotype and abundance was performed using ImmunoSEQ Analyzer (Adaptive Biotechnologies).
Integration site analysis
Quantitative lentivector integration site analysis was performed using linker-mediated PCR as described previously.12 Briefly, patient DNA is randomly sheared, end-repaired, and ligated to a linker. The integration site is amplified with 1 primer specific to the lentivector LTR and another primer specific to the linker. The amplified product is subjected to high-throughput Illumina Sequencing. Integration sites in the sample are identified and quantified by corresponding sequence reads for clonality analysis. Further confirmation of quantification of CBL integration was performed using a ddPCR assay specific for the CBL-lentivector junction with 1 primer in the CBL gene and the other primer in the vector LTR. Copy number per cell of the CBL-lentivector junction was normalized to the input DNA with an assay for the human MKL2 gene as the reference (CBL gene-specific primer: 5′CCCATTTTCTTATTGAGTGGTTTATC3′; lentivector LTR-specific primer: 5′CCCACTGCTTAAGCCTCAATA3′; lentivector probe: 5′/56-FAM/AAATCTCTA/ZEN/GCAGTGGCGCCCG/3IABkFQ/3′; MKL2 Fwd primer: 5′AGATCAGAAGGGTGAGAAGAATG3′; MKL2 Rev primer: 5′GGATGGTCTGGTAGTTGTAGTG3′; MKL2 probe: 5′-/5HEX/TGTTCCTGC/ZEN/AACTGCAGATCCTGA/3IABkFQ/-3′). Primers and probe were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). ddPCR assay was performed using BioRad Droplet Digital PCR system (Hercules, CA).
Results and discussion
Insertional mutagenesis has been a concern for human gene therapies using retroviral vectors, particularly when performed on hematopoietic stem cells (HSCs). The 2 types of retroviral vectors most widely used are γ-retroviral vectors (based on the backbone of γ-retrovirus, such as murine leukemia virus [MLV]) and lentiviral vectors (based on the backbone of a lentivirus such as HIV 1). In the gene therapy trials for X-linked severe combined immunodeficiency syndromes, insertion of the murine leukemia virus-based vector carrying common cytokine receptor γ gene IL2RC in HSCs resulted in a series of immature T-cell leukemias.13 Integration site analysis demonstrated that these leukemias were associated with insertion of the transgene in the LMO2 locus, which presumably caused an activation of this cellular oncogene by the promoter/enhancer elements contained in the γ-retroviral vector. The insertional mutagenesis potential of vectors may be related to the intrinsic integration preference of the retrovirus used. γ-retrovirus or γ-retroviral vector preferentially integrate near gene regulatory sites such as enhancers and transcription start sites, potentially increasing the possibility of causing dysregulation of target genes.14-16 Lentivirus or lentiviral vectors do not have the same predilection for integration into such regulatory regions, potentially reducing the possibility of gene dysregulation. However, lentiviral vectors do preferentially integrate into actively transcribed genes, which still poses potential risk of mutagenesis.14,17 This concern is enhanced by the demonstration that insertion of an HIV provirus within specific regions of the MKL2 and BACH2 genes in HIV-infected patients can cause a clonal expansion.18,19 Gene therapy trials using lentiviral vectors also incorporate additional safety features such as self-inactivation that removes part of the regulatory region in the 3LTR.20 Although adverse events associated with insertional mutagenesis have not been reported in gene therapy trials involving lentiviral vectors,21 vector-mediated clonal expansion has been reported for β-thalassemia gene therapy.22 Thus, long-term monitoring is mandated by regulatory agencies to assess for this potential complication in gene therapy trials. Viral vector-based CAR T-cell therapy uses differentiated T cells, which may reduce the risks associated with insertional mutagenesis relative to gene therapy using HSCs. Furthermore, replication competent retrovirus and replication competent lentivirus testing from multiple CAR T-cell trials has not demonstrated the presence of replicating virus in CAR T-cell products manufactured with either γ-retrovirus or lentivirus.21,23
In our report, both TCR sequencing and integration site analysis allowed for comparison of the clonal composition of the T-cell population at the 2 peaks of CAR T-cell expansion in this patient. The infused CAR T cells and the initial peak of CAR T-cell expansion were largely polyclonal. During the second peak of CAR T-cell expansion, 1 clone containing an insertion of the CAR vector in the CBL gene constituted approximately one-half of total WBCs. This clone was not detectable in the infused CAR T product (the ddPCR detection sensitivity for this clone was at 1/10 000 cells with the DNA input we used), but became detectable at a low level (2.6%) as early as day +7 during the first CAR T-cell expansion and remained below 1% (but detectable) at days +15 and +30. This single clone increased dramatically from day +51 to +53 during a second CAR T-cell expansion without associated CRS symptoms.
One possibility is that the large T-cell clone was associated with antigen stimulation through the endogenous TCR, although the patient tested negative for Epstein-Barr virus or cytomegalovirus infection, suggesting that T-cell clone was not expanded via the endogenous TCR by reactivation of these 2 viruses. The TCR sequence of this clone does not match known TCR sequences to other antigens in the VDJ database (https://vdjdb.cdr3.net). A second possibility is that the clone was expanded via the CAR in the setting of residual lymphomatous disease despite absence of leukemia in the bone marrow. Finally, and not mutually exclusive of stimulation through the CAR or TCR, is that the provirus integration into the CBL gene contributed to the clonal expansion. A truncated form of the CBL gene, v-Cbl (named for Casitas B-Lineage Lymphoma), was first discovered as a viral oncogene in mice. The v-Cbl segment comprises the tyrosine kinase binding domain portion of the cellular counter-part c-Cbl (official gene name CBL). The CBL gene encodes a E3 ubiquitin-protein ligase and has closely related homolog CBL-B, and CBL-C. CBL and CBL-B have both been described in the regulation of mature TCR signaling and loss of CBL-B reduces the threshold for T-cell activation and dependence on costimulation.24 CBL has similar effects in developing thymocytes.25 Structurally and functionally, CBL and CBL-B are closely related and activity in different stages of T-cell development relates in part to differential expression. Knock down of CBL and CBL-B has also showed an increased therapeutic activity of adoptively transferred T cells against tumors.26-29 Recent data have demonstrated that a lack of CBL-B results in diminished T-cell sensitivity to PD-1-mediated inhibition.30 It is unlikely that the integration caused a complete loss of function of CBL because the integration occurred not in the exon, but rather in the second intron of only 1 CBL allele. Because T cells from mice with a single intact CBLB allele do not demonstrate enhanced activity in tumor models,31 it is possible that the single insertion resulted in a dominant-negative effect or interference with the normal CBL and/or CBLB function, thus contributing to the hyperexpansion in response to a small amount of antigen. Further mechanistic studies are under way.
Recently, Fraietta et al reported a case of clonal CAR T-cell expansion in the context of anti-CD19 CAR T-cell therapy, which involved the integration of a lentiviral vector in the TET2 gene.8 TET2 encodes a methylcytosine dioxygenase that is involved in DNA demethylation and is a key regulator of hematopoiesis. The disruption of 1 TET2 allele by the lentivector through alternative splicing into the vector, in combination with the hypomorphic mutation in the other TET2 allele carried by the patient, resulted in a clonal expansion and led, at the peak of expansion, to 94% of the CAR T cells originating from the clone containing the TET2 insertion. However, in our patient, no mutations were found in either allele of the CBL gene. Our results raise the possibility that specific integration of the CAR into other host genes involved in T-cell activation could enhance expansion and/or function of CAR T cells.
In conclusion, we report, to the best of our knowledge, on the first case of clonal expansion seen with CD22 CAR T-cell targeted therapy associated with lentivector integration in a gene known to regulate T-cell responses. Only 2 cases of CAR T-cell associated clonal expansion, 1 involving an insertion in TET2, and our case involving CBL, have been reported. However, in both cases, the clonal expansion was clinically apparent and not subtle, suggesting that current clinical practices for monitoring adverse events are likely adequate. Our case differs from the prior case in that our patient had significant expansion without concurrent signs of CRS. In both cases, the delayed expansion occurred in the presence of ongoing residual disease, suggesting that there may be clones that have a lower threshold for activation, in which antigen may potentially trigger a very large expansion. Whether, and to what extent, the integrated lentiviral vector affected the expression of expression of CBL and/or CBLB remains to be determined but analyses such as this may provide further insights into the regulation of T-cell signaling pathways and the optimization of genetically modified T-cell therapies.
Acknowledgments
The authors thank the patient, referring medical team, and National Cancer Institute/Center for Cancer Research clinical teams, and protocol research teams support. The subject provided consent to publication of this finding.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (HHSN261200800001E) (X.W.).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The subject was made aware of the findings and consented to publication of this manuscript.
Authorship
Contribution: N.N.S., T.J.F., and X.W. all contributed to the first version of this manuscript; H.Q. evaluated for chimeric antigen receptor persistence by polymerase chain reaction; L.S., S.G., S.L., S.H.H., and X.W. performed the ImmunoSeq analysis, integration site analysis, and Droplet Digital polymerase chain reaction analysis; N.N.S., B.Y., and H.S. contributed to the coordination and care of the patient reported in this manuscript; M.S.-S. and C.Y. conducted all the clinical flow cytometry analysis for minimal residual disease and diagnostic evaluation as well as chimeric antigen receptor detection by flow cytometry; N.N.S. and T.J.F. conducted this clinical trial; M.R. conducted the molecular studies that led to the identification of a clonal expansion; M.A.A. provided clinical input, figure contribution, and diagnostic assessment by 18fluorodeoxyglucose positron emission tomography imaging; and all authors reviewed the manuscript and provided intellectual input and made critical revisions leading to the final submission of this manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nirali N. Shah, National Institutes of Health, Building 10/CRC, Room 1-5750, 10 Center Dr, Bethesda, MD 20892; e-mail: shahnn@mail.nih.gov.

