

Key Points
Gas6 serum levels were increased in HSCT patients with grade II-IV acute GVHD.
The Gas6-Mer axis contributes to endothelial dysfunction in a common pathology between TA-TMA and GVHD.

Abstract
Endothelial dysfunction in the early phases of hematopoietic stem cell transplantation (HSCT) contributes to a common pathology between transplant-associated thrombotic microangiopathy (TA-TMA) and graft-versus-host disease (GVHD), which are serious complications of HSCT. Growth arrest-specific (Gas) 6 structurally belongs to the family of plasma vitamin K-dependent proteins working as a cofactor for activated protein C, and has growth factor-like properties through its interaction with receptor tyrosine kinases of the TAM family: Tyro3, Axl, and Mer. Serum Gas6 levels were significantly increased in HSCT patients with grade II to IV acute GVHD (aGVHD), and Gas6 and Mer expression levels were upregulated in aGVHD lesions of the large intestine and skin. The increased serum Gas6 levels were also correlated with elevated lactate dehydrogenase, d-dimer, and plasmin inhibitor complex values in HSCT patients with aGVHD. In human umbilical vein endothelial cells (ECs), exogenous Gas6 or the exposure of sera isolated from patients with grade III aGVHD to ECs induced the downregulation of thrombomodulin and the upregulation of PAI-1, as well as the upregulation of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1, which were inhibited by UNC2250, a selective Mer tyrosine kinase inhibitor. In mouse HSCT models, we observed hepatic GVHD with hepatocellular apoptosis, necrosis, and fibrosis, as well as TA-TMA, which is characterized pathologically by thrombosis formation in the microvasculature of the liver and kidney. Of note, intravenous administration of UNC2250 markedly suppressed GVHD and TA-TMA in these mouse HSCT models. Our findings suggest that the Gas6-Mer axis is a promising target for TA-TMA after GVHD.
Introduction
Hematopoietic stem cell transplantation (HSCT) is recognized as the definitive therapy for hematological malignancies and disorders.1,2 Graft-versus-host disease (GVHD) and transplant-associated thrombotic microangiopathy (TA-TMA) are crucial complications associated with significant morbidity and mortality, and management of these complications contributes to an increase in the success rate of HSCT.3,4 Pro-inflammatory cytokines such as interleukin 6 (IL-6), IL-1β, interferon-γ, and tumor necrosis factor (TNF)–α play a critical role in the pathogenesis of GVHD,5-7 and activated donor T lymphocytes develop a burst of cytokine production and damage-associated molecular patterns, which are derived from tissue-specific destruction of GVHD target organs.5,7,8 TA-TMA induced by endothelial dysfunction in the early phase of HSCT results in microangiopathic hemolytic anemia, as well as platelet activation and consumption, leading to the formation of platelet-rich thrombi in the microcirculation.3,9 Higher acute GVHD (aGVHD) grade and steroid-refractory GVHD are known risk factors for the development of TA-TMA. Other risk factors include high-dose chemotherapy, radiation therapy, HLA mismatch, the use of calcineurin inhibitors, cytomegaloviral infection, age, and sex.9,10 In addition, patients with aGVHD are at a fourfold risk of developing TA-TMA when compared with patients without aGVHD.11
Endothelial function, which maintains anti-inflammatory and antithrombotic surfaces, orchestrates inflammatory and thrombotic responses in the vascular wall.12,13 Endothelial dysfunction caused by the toxic effects of high-dose chemotherapy, radiation therapy, and exposure to calcineurin inhibitors is an important early event in the pathogenesis of HSCT-associated complications.5-7 Host endothelial dysfunction is mediated by cytotoxic T lymphocytes and/or pro-inflammatory cytokines, and substantial blood vessel loss may lead to impaired blood perfusion and tissue fibrosis, the characteristic lesion of GVHD.14,15 Plasma levels of endothelial cell (EC) activation markers such as adhesion molecules including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in HSCT recipients developing GVHD, as well as TA-TMA,16 and thrombomodulin (TM) and plasminogen activator inhibitor (PAI-1) have also been reported to be indicators of endothelial damage in the early phase of HSCT-related complications.17,18 The increased numbers of circulating ECs have also been reported to be a new marker to monitor microvascular endothelial dysfunction after HSCT.19 TA-TMA is a multifactorial disorder, which can be triggered by systemic microvascular endothelial dysfunction with platelet activation and consumption, as well as coagulation factors after HSCT.3,20 The pathological characteristics of TA-TMA typically show intravascular platelet-rich thrombus, microangiopathic hemolytic anemia, and thrombocytopenia caused by microvascular endothelial dysfunction after HSCT.3,21 Endothelial dysfunction after HSCT is a critical mechanism that contributes to the clinical overlap between GVHD and TA-TMA.9,10,19 However, treatment of these complications has not yet been well established.
Growth arrest-specific (Gas) 6 structurally belongs to the family of plasma vitamin K-dependent proteins and has growth factor–like properties through its interaction with receptor tyrosine kinases of the TAM family: Tyro3, Axl, and Mer.22-24 Gas6, protein S, and the TAM receptors affects primary hemostasis and coagulation, and display anti- or pro-inflammatory effects, depending on the cell type, and even on the receptor.23,24 Plasma Gas6 levels correlate with systemic lupus erythematosus disease activity,23 and some investigators have reported that Gas6-related signaling pathways may be critical for the progression mechanisms in hematological malignancies.24-27 We previously reported new evidence that autocrine and paracrine stimulation of Gas6 contributes to the pathogenesis of multiple myeloma.27 Gas6 regulates platelet activation, the levels of platelet P-selectin, and subsequent interactions with leukocytes and ECs during inflammation.28-30 Gas6 promotes the recruitment of inflammatory monocytes during deep venous thrombosis.29,30 Inhibition of Gas6 or its receptors provides antithrombotic activity in the absence of increased bleeding, and therefore presents an attractive anticoagulant drug target.30 Recently, the lack of Gas6 in recipient mice has been reported to alleviate hepatic GVHD, suggesting that Gas6 is a potential non-immunosuppressive target to minimize hepatic GVHD in patients receiving HSCT.31 However, the detailed mechanisms have not yet been fully elucidated.
In the present study, we hypothesized that the receptor tyrosine kinase Mer activated by Gas6 may contribute to clinical overlap between GVHD and TA-TMA undergoing HSCT. We suggest that the Gas6-Mer axis can potentially be an attractive therapeutic target for the treatment of endothelial dysfunction involved in GVHD and TA-TMA.
Methods
Patients
Peripheral blood samples were obtained from 14 consecutive patients (mean age, 50 years: 47 years for males and 60 years for females) who underwent HSCT in their first complete remission between 2017 and 2018 at Fukushima Medical University Hospital (Table 1). This study was performed after obtaining approval from the Ethic Committee of Fukushima Medical University, which is guided by local policy, national law, and the World Health Association Declaration of Helsinki. We used the sera isolated from 4 patients with grade III intestinal aGVHD for in vitro experiments at 21 to 35 days after HSCT. We carried out all in vitro experiments using the sera, which were collected using the same method. Organ staging of aGVHD is based on the degree of 3 main target organ damages (skin, liver, and gut), and its overall grading is then determined using the modified Seattle Glucksberg criteria.32,33 For patients with chronic GVHD (cGVHD), the severity criteria recommended by the National Institutes of Health consensus development project were employed.34 The number of organs involved and the severity of the disease in these organs was dictated by the global summary score to define the disease as mild, moderate, or severe. Mild disease indicates the involvement of 1 or 2 organs, each with a score of 1. Moderate disease indicates moderate lesions with a score of 2, 3 or more organs with a score of 2 in any organ, or lung involvement with a score of 1. Severe disease is a score of 2 in the lung or a score of 3 in any other organ.34,35 The published diagnostic criteria for TA-TMA include at least 2 schistocytes per high-power field in peripheral blood, increased lactate dehydrogenase (LDH), thrombocytopenia lower than 50×109/L, or a 50% decrease in platelet count, decreased hemoglobin, negative Coombs test, and decreased haptoglobin.36 The clinical manifestations of engraftment syndrome (ES) have been commonly reported to be fever, rash, and noncardiogenic pulmonary edema. Further, ES occurs within 96 hours of engraftment (neutrophil count ≥500/μL for 2 consecutive days). A diagnosis of ES is established by the presence of all 3 major criteria, or of 2 major criteria and 1 or more minor criteria.37
Clinical parameters for HSCT patients with or without GVHD
| Case | Age, y | Sex | Disease | Donor | Conditioning regimens | Immunosuppressant drugs | GVHD | ES | GVHD therapy | Steroid-refractory GVHD |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 35 | M | CLL | BM | CY/TBI 12Gy | Tacrolimus short course MTX | aGVHD grade II (skin stage 3), severe cGVHD (lung score of 3) | + | PSL tacrolimus | + |
| 2 | 59 | M | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade III (skin stage 1, gut stage 4) | + | PSL tacrolimus hMSC | + |
| 3 | 64 | F | T-ALL | CBT | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade II (skin stage 3) | + | PSL tacrolimus | + |
| 4 | 36 | M | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade I (skin stage 1) | − | Topical agents | − |
| 5 | 61 | M | AML | BM | Flu/Mel/Bu | Tacrolimus short course MTX | aGVHD grade III (skin stage 1, gut stage 3) | − | PSL | − |
| 6 | 47 | M | ALL | BM | Flu/Mel/Bu | Tacrolimus short course MTX | — | − | − | − |
| 7 | 62 | F | ALL | BM | Flu/Mel/Bu | Tacrolimus short course MTX | aGVHD grade III (skin stage 1, gut stage 3) | + | PSL tacrolimus | + |
| 8 | 52 | M | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | — | − | − | − |
| 9 | 20 | M | AML | Haplo-PB | Flu/Bu/TBI 4Gy | Tacrolimus MMF, PTCy | aGVHD grade IV (skin stage 1, liver stage 4) | − | PSL tacrolimus | + |
| 10 | 58 | M | MDS | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade I (skin stage 1) | − | Topical agents | − |
| 11 | 64 | M | AML | Haplo-PB | Flu/Bu/TBI 4Gy | Tacrolimus MMF, PTCy | aGVHD grade II (skin stage 3) Severe cGVHD (lung score of 3) | + | PSL tacrolimus | + |
| 12 | 53 | M | MDS | CBT | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | — | − | − | − |
| 13 | 37 | M | MDS | BM | Bu/CY | ATG, tacrolimus short course MTX | aGVHD grade I (skin stage 1) | + | Topical agents | − |
| 14 | 54 | F | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | — | − | − | − |
| Case | Age, y | Sex | Disease | Donor | Conditioning regimens | Immunosuppressant drugs | GVHD | ES | GVHD therapy | Steroid-refractory GVHD |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 35 | M | CLL | BM | CY/TBI 12Gy | Tacrolimus short course MTX | aGVHD grade II (skin stage 3), severe cGVHD (lung score of 3) | + | PSL tacrolimus | + |
| 2 | 59 | M | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade III (skin stage 1, gut stage 4) | + | PSL tacrolimus hMSC | + |
| 3 | 64 | F | T-ALL | CBT | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade II (skin stage 3) | + | PSL tacrolimus | + |
| 4 | 36 | M | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade I (skin stage 1) | − | Topical agents | − |
| 5 | 61 | M | AML | BM | Flu/Mel/Bu | Tacrolimus short course MTX | aGVHD grade III (skin stage 1, gut stage 3) | − | PSL | − |
| 6 | 47 | M | ALL | BM | Flu/Mel/Bu | Tacrolimus short course MTX | — | − | − | − |
| 7 | 62 | F | ALL | BM | Flu/Mel/Bu | Tacrolimus short course MTX | aGVHD grade III (skin stage 1, gut stage 3) | + | PSL tacrolimus | + |
| 8 | 52 | M | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | — | − | − | − |
| 9 | 20 | M | AML | Haplo-PB | Flu/Bu/TBI 4Gy | Tacrolimus MMF, PTCy | aGVHD grade IV (skin stage 1, liver stage 4) | − | PSL tacrolimus | + |
| 10 | 58 | M | MDS | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | aGVHD grade I (skin stage 1) | − | Topical agents | − |
| 11 | 64 | M | AML | Haplo-PB | Flu/Bu/TBI 4Gy | Tacrolimus MMF, PTCy | aGVHD grade II (skin stage 3) Severe cGVHD (lung score of 3) | + | PSL tacrolimus | + |
| 12 | 53 | M | MDS | CBT | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | — | − | − | − |
| 13 | 37 | M | MDS | BM | Bu/CY | ATG, tacrolimus short course MTX | aGVHD grade I (skin stage 1) | + | Topical agents | − |
| 14 | 54 | F | AML | BM | Flu/Bu/TBI 4Gy | Tacrolimus short course MTX | — | − | − | − |
ATG, antithymocyte globulin; ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; Bu, busulfan; CBT, cord blood transplantation; CLL, chronic lymphocytic leukemia; CY, cyclophosphamide; ES, engraftment syndrome; F, female; Flu, fludarabine; GVHD, graft-versus-host disease; Gy, Gray; hMSC, human mesenchymal stem cell; M, male; Mel, melphalan; MDS, myelodysplastic syndrome; MMF, mycophenolate mofetil; MTX, methotrexate; PB, peripheral blood; PSL, prednisolone; PTCy, posttransplant high-dose cyclophosphamide; TBI, total body irradiation.
Enzyme-linked immunosorbent assay
The levels of Gas6 protein were assessed in human or mouse plasma, using a commercially available Gas6 enzyme-linked immunosorbent assay (ELISA) kit (Assay Biotechnology) in accordance with the manufacturer’s instructions. The reaction was stopped with 2N sulfuric acid, and the absorbance at 450 nm was read using a microplate reader (Molecular Devices), with a reference wavelength set at 570 nm.
Cell culture and reagents
Human umbilical vein ECs were cultured according to the suppliers’ instructions (Clonetics Inc., Walkersville, MD; Sanko Junyaku Co., Ltd., Tokyo, Japan). They were used for all experiments after 5 to 10 passages. Recombinant human Gas6 was purchased from R&D Systems, Inc. (Minneapolis, MN), and UNC2250, a Mer-selective small molecular inhibitor, was obtained from Axon Medchem LLC. UNC2250 is a potent and selective Mer inhibitor with a 50% inhibitory concentration of 9.8 nM and an approximate 60- and 160-fold selectivity over the closely related kinases Axl and Tyro3, respectively, and also targets other serine/threonine such as the epidermal growth factor receptor.38
Flow cytometry
The cells were treated with an anti-Gas6 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA), conjugated to an anti-FITC secondary antibody (Santa Cruz) and then stained with an anti-CD3-PE antibody (Santa Cruz), an anti-CD14-PE antibody (Santa Cruz), and anti-CD19-PE antibody (Santa Cruz), using flow cytometry (BD Biosciences). Finally, they were analyzed by flow cytometry.
Immunostaining
The specimens were stained with hematoxylin and eosin and examined under microscopy. After rinsing with phosphate-buffered saline (PBS), the sections were embedded in paraffin for immunostaining. In addition, the sections were prepared, deparaffinized, and autoclaved at 121°C for 10 minutes in 10 mM Tris⋅HCl buffer (pH 9.0) as an antigen retrieval procedure. The sections were then blocked and incubated with a goat polyclonal antibody against Gas6 (dilution 1:100, Santa Cruz) or mouse monoclonal antibodies against Mer (dilution 1:250, Santa Cruz), Axl, and Tyro3 at 4°C for 2 days. We used anti-mouse Alexa 488 and anti-goat Alexa 594 (Molecular Probes, Eugene, OR), which were reacted for 60 minutes for fluorescent immunostaining. Stained tissues were stored in the dark until they were analyzed by a confocal microscope (Keyence, IL).
Polymerase chain reaction
RNA was isolated with an Absolutely RNA Miniprep Kit (Agilent Technologies, Cedar Creek, TX). Genomic DNA was digested with DNAse I (Agilent Technologies), and RNA was then reverse transcribed with SuperScript III First-Strand (Invitrogen). Quantitative polymerase chain reaction (PCR) was performed using Power SYBR Green PCR master mix and the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). The threshold cycle (Ct) was calculated using threshold cycles for the target genes (Gas6, Mer, TNF-α, IL-1β, and β-actin). These primers were purchased from Takara Bio (Shiga, Japan).
Bone marrow transplantation
Recipient BALB/c mice received a single dose of 7 Gy total body irradiation (137Cs source, 129 cGy/minute) from Softex M-150 WE (Softex, Tokyo, Japan). On the day of transplantation, the donor mice (C57BL/6 for allogeneic transplantation; BALB/c for syngeneic transplantation) were killed by cervical dislocation. Their femora were removed aseptically, and bone marrow (BM) was removed from the femoral shaft via insertion of a 25-gauge needle at the proximal end. Irradiated BALB/c mice received a single injection through the tail vein of 0.25 mL RPMI 1640 medium containing nucleated BM cells and nucleated spleen cells.
Western blotting
Rabbit polyclonal antibodies to ICAM-1 (Santa Cruz) and VCAM-1 (Santa Cruz) were diluted 1:250 in PBS with 5% bovine serum albumin. We used anti-TM antibody (dilution 1:250, Cell Signaling Technology, Inc.) and anti-PAI-1 antibody (dilution 1:250, Santa Cruz). Anti-β-actin antibody (dilution 1:1000, Santa Cruz) was used as a loading control. We used anti-phosho-Mer antibody (dilution 1:250; Abcam, Cambridge, United Kingdom) and anti-phosho-Axl antibody (dilution 1:250; Assay Biotechnology). Phosphorylation of Tyro3 was examined using anti-phosphotyrosine (dilution 1:250; MilliporeSigma) after immunoprecipitation with anti-Tyro3 antibody (Santa Cruz). Individual bands were quantitatively analyzed by the NIH Image Program (ImageJ).
Platelet aggregation
Platelet aggregation was performed in human citrated platelet-rich plasma. Human platelet aggregation in response to 2.5 μg/mL collagen or 5 μmol/L adenosine 5′-diphosphate (ADP) was determined by light transmittance aggregometry (MC Medical, Inc., Tokyo, Japan), as previously described.39 Platelet-poor plasma was used as a negative control. Platelet aggregation was defined as the percentage of maximum platelet aggregation.
Terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate nick end labeling assay
Terminal deoxynucleotidyltransferase buffer containing 0.3 U/mL terminal deoxynucleotidyltransferase (Boehringer, Mannheim, Germany) and 10 mM biotinylated 16-deoxyuridine triphosphate (Boehringer) was added to the cells, which were then incubated at 37°C for 60 minutes. After incubation with 5% bovine serum albumin in PBS for 1 hour at room temperature to block nonspecific binding, the cells were incubated with horseradish peroxidase-labeled goat anti-biotin (1:100) diluted with 5% bovine serum albumin in PBS for 2 hours at room temperature, and then washed in PBS. The horseradish peroxidase sites were visualized by H2O2 and 3,3'-diaminobenzidine for 10 minutes. Terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay images were stored in a computer, and the intensity of EC apoptosis staining was quantitatively analyzed by the NIH Image Program (ImageJ).
Statistical analysis
Statistical analyses were performed using analysis of variance with Scheffé’s post hoc test when appropriate. P < .05 was considered statistically significant. Data are expressed as means ± standard deviation (SD).
Results
Serum Gas6 levels in patients with GVHD after HSCT
We first performed ELISA for the detection of serum Gas6 levels to investigate whether Gas6 assumes a key role in early complications after HSCT. As shown in Figure 1A, the serum Gas6 levels were significantly increased in HSCT patients at 21 to 35 days after stem cell engraftment, as determined by a human Gas6 ELISA kit (Figure 1A). The serum Gas6 levels were also markedly increased in the HSCT patients with grade II to IV aGVHD, compared with those with grade 0 to I aGVHD (Figure 1B). Early complications such as ES caused by systemic endothelial dysfunction were defined as the clinical syndrome of fever, skin rash, noncardiogenic pulmonary edema, weight gain, and liver and renal dysfunction, accompanied with neutrophil recovery after HSCT.37 ELISA also showed that at 3 weeks after HSCT, the serum Gas6 levels were significantly increased in the group with ES (n = 6) compared with the group without ES (n = 8; Figure 1C). Flow cytometric analysis showed that Gas6 protein levels were increased in the CD3-positive lymphocytes and CD14-positive monocytes, which were obtained from the blood of patients with aGVHD (Figure 1Ea-c). Meanwhile, little to no Gas6 protein was detectable in the CD3-positive lymphocytes or CD14-positive monocytes in the blood of those without aGVHD (Figure 1Da-c). The clinical signs of TA-TMA have been reported to be elevated blood LDH, proteinuria, renal dysfunction, thrombocytopenia, and hypertension.3,36,40 Our analysis results showed that LDH, resistance to platelet transfusion, creatinine levels, and thrombin-antithrombin complex values were significantly increased in the patients with grade II to IV aGVHD compared with those with grade 0 to I aGVHD after HSCT (Figure 1Fa-d). In addition, we showed that the serum Gas6 levels were correlated with LDH, d-dimer, and plasmin-alpha2 plasmin inhibitor complex values in 3 representative cases with grade II to III aGVHD after HSCT (Figure 1G). These results suggest that higher Gas6 levels may contribute to the pathogenesis of early-stage complications such as GVHD and TA-TMA.

The levels of serum Gas6 in patients with aGVHD or engraftment syndrome after HSCT. (A) The levels of serum Gas6 in patients within 6 weeks after HSCT were quantified by a human Gas6 ELISA kit. Data are expressed as mean ± SD (n = 14). *P < .05. (B) The levels of serum Gas6 were quantified by a human Gas6 ELISA kit in patients with grade 0 to I aGVHD (n = 7) and grade II-IV aGVHD (n = 7). Data are expressed as mean ± SD. *P < .05. (C) The levels of serum Gas6 in the patients with ES (n = 6) were significantly increased in comparison with those without ES (n = 8), using a human Gas6 ELISA kit. Data are expressed as mean ± SD. *P < .05. (Da-c) Gas6 protein was examined on the surface of CD3-positive lymphocytes, CD14-positive monocytes, and CD19-positive lymphocytes in the peripheral blood mononuclear cells of the patients without aGVHD, using flow cytometry data analysis. Representative data are from 3 independent experiments. (Ea-c) Our flow cytometry data analysis shows that Gas6 protein was examined on the surface of CD3-positive lymphocytes, CD14-positive monocytes, and CD19-positive lymphocytes in peripheral blood mononuclear cells of patients with aGVHD. Representative data are from 3 independent experiments. (Fa-d) LDH, creatinine levels, resistance to platelet transfusion, and thrombin-antithrombin complex values were significantly increased in the patients with grade II to IV aGVHD (n = 7) compared with those with grade 0 to I aGVHD (n = 7) at 3, 4, and 5 weeks after HSCT. Data are expressed as mean ± SD. *P < .05. (G) The levels of serum Gas6 correlated with blood LDH, d-dimer, and plasmin-alpha2 plasmin inhibitor complex (PIC) values in 3 representative cases with grade II to III aGVHD after HSCT.
The levels of serum Gas6 in patients with aGVHD or engraftment syndrome after HSCT. (A) The levels of serum Gas6 in patients within 6 weeks after HSCT were quantified by a human Gas6 ELISA kit. Data are expressed as mean ± SD (n = 14). *P < .05. (B) The levels of serum Gas6 were quantified by a human Gas6 ELISA kit in patients with grade 0 to I aGVHD (n = 7) and grade II-IV aGVHD (n = 7). Data are expressed as mean ± SD. *P < .05. (C) The levels of serum Gas6 in the patients with ES (n = 6) were significantly increased in comparison with those without ES (n = 8), using a human Gas6 ELISA kit. Data are expressed as mean ± SD. *P < .05. (Da-c) Gas6 protein was examined on the surface of CD3-positive lymphocytes, CD14-positive monocytes, and CD19-positive lymphocytes in the peripheral blood mononuclear cells of the patients without aGVHD, using flow cytometry data analysis. Representative data are from 3 independent experiments. (Ea-c) Our flow cytometry data analysis shows that Gas6 protein was examined on the surface of CD3-positive lymphocytes, CD14-positive monocytes, and CD19-positive lymphocytes in peripheral blood mononuclear cells of patients with aGVHD. Representative data are from 3 independent experiments. (Fa-d) LDH, creatinine levels, resistance to platelet transfusion, and thrombin-antithrombin complex values were significantly increased in the patients with grade II to IV aGVHD (n = 7) compared with those with grade 0 to I aGVHD (n = 7) at 3, 4, and 5 weeks after HSCT. Data are expressed as mean ± SD. *P < .05. (G) The levels of serum Gas6 correlated with blood LDH, d-dimer, and plasmin-alpha2 plasmin inhibitor complex (PIC) values in 3 representative cases with grade II to III aGVHD after HSCT.
Expression of Gas6 and TAM receptors in aGVHD lesions of the large intestine and skin
To identify which of the receptor tyrosine kinases Mer, Axl, and Tyro3 contributes to the Gas6-mediated signaling pathway, immunostaining of aGVHD lesions of the large intestine and skin was performed. Our analysis of the immunohistochemistry (IHC) results show that little or no Gas6, Mer, Axl, or Tyro3 expression was observed in the large intestine of patients with negative histology for aGVHD (Figure 2Aa-d). In addition, our IHC results showed that infiltrating CD3-positive T lymphocytes were markedly increased in aGVHD lesions of the large intestine (Figure 2Ba-b). IHC analysis of aGVHD lesions of the large intestine and skin showed that there was a positive correlation between the high expression of Gas6 (Figure 2Bc-d,Ca) and that of Mer (Figure 2Be-f,Cb), whereas little to no expression of Axl or Tyro3 was detectable in the aGVHD lesions (Figure 2Bg-h,Cc-d). In addition, double immunofluorescence staining showed that Gas6 bonded to Mer in the aGVHD lesions (Figure 2Bi-k,Ce-g). These findings indicate that Mer may be a key receptor for mediating the Gas6-related signaling pathway in the pathogenesis of aGVHD.

Gas6 and Mer expression in aGVHD lesions of the large intestine and skin. (Aa-d) IHC of Gas6, Mer, Axl, and Tyro3 in the intestinal tissues of patients without aGVHD was performed. Analysis of the IHC results indicated that little to no expression of Gas6, Mer, Axl, or Tyro3 was observed in the large intestine of patients with negative histology for aGVHD. Scale bars represent 10 µm. Original magnification ×200. (Ba-b) CD3-positive infiltrating T lymphocytes were markedly increased in aGVHD lesions of the large intestine. Scale bars represent 10 µm. Original magnification ×200. (Bc-h) Gas6 and Mer were significantly upregulated in the aGVHD lesions of the large intestine, whereas little to no Axl or Tyro3 was detectable. Immunohistochemical stain for Gas6, Mer, Axl, or Tyro3. Arrows indicate Gas6 and Mer expression in aGVHD lesions of the large intestine. Scale bars represent 10 µm. Original magnification ×200. (Bi-k) Fluorescent IHC of Gas6 and Mer in aGVHD lesions of the large intestine was performed. Scale bars represent 10 µm. Original magnification ×200. (Bl) The statistical analysis revealed that the expression of Gas6 and Mer was increased in the aGVHD lesions of the large intestine compared with patients with no such lesions. We measured the intensity of Gas6 and Mer expression levels by color densitometry, using image J. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. **P < .01, *P < .05. (Ca-d) IHC of Gas6, Mer, Axl, and Tyro3 in the aGVHD lesions of the skin was performed. Scale bars represent 50 µm. Original magnification ×200. (Ce-g) Fluorescent IHC of Gas6 and Mer in aGVHD lesions of the skin was performed. Scale bars represent 50 µm. Original magnification ×200.
Gas6 and Mer expression in aGVHD lesions of the large intestine and skin. (Aa-d) IHC of Gas6, Mer, Axl, and Tyro3 in the intestinal tissues of patients without aGVHD was performed. Analysis of the IHC results indicated that little to no expression of Gas6, Mer, Axl, or Tyro3 was observed in the large intestine of patients with negative histology for aGVHD. Scale bars represent 10 µm. Original magnification ×200. (Ba-b) CD3-positive infiltrating T lymphocytes were markedly increased in aGVHD lesions of the large intestine. Scale bars represent 10 µm. Original magnification ×200. (Bc-h) Gas6 and Mer were significantly upregulated in the aGVHD lesions of the large intestine, whereas little to no Axl or Tyro3 was detectable. Immunohistochemical stain for Gas6, Mer, Axl, or Tyro3. Arrows indicate Gas6 and Mer expression in aGVHD lesions of the large intestine. Scale bars represent 10 µm. Original magnification ×200. (Bi-k) Fluorescent IHC of Gas6 and Mer in aGVHD lesions of the large intestine was performed. Scale bars represent 10 µm. Original magnification ×200. (Bl) The statistical analysis revealed that the expression of Gas6 and Mer was increased in the aGVHD lesions of the large intestine compared with patients with no such lesions. We measured the intensity of Gas6 and Mer expression levels by color densitometry, using image J. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. **P < .01, *P < .05. (Ca-d) IHC of Gas6, Mer, Axl, and Tyro3 in the aGVHD lesions of the skin was performed. Scale bars represent 50 µm. Original magnification ×200. (Ce-g) Fluorescent IHC of Gas6 and Mer in aGVHD lesions of the skin was performed. Scale bars represent 50 µm. Original magnification ×200.
Effects of Mer inhibition on ICAM-1 and VCAM-1 upregulation induced by Gas6 or the exposure of sera isolated from patients with grade III aGVHD in ECs
Endothelial dysfunction after HSCT is induced by a complex process of engraftment involving high-dose chemoradiotherapy, radiation, immunosuppressive drugs, and pro-inflammatory cytokines.6,7,41 ICAM-1 and VCAM-1 are involved in cell-adhesive events that trigger multiple cell-signaling pathways, endothelial dysfunction, inflammation, hypercoagulation, and cell apoptosis.16,42 Serum soluble VCAM-1 is significantly elevated in patients with GVHD and/or TA-TMA.16 Gas6 regulates thrombin-induced expression of VCAM-1 in ECs by promoting adhesion of bone marrow mononuclear cells on ECs.43 Our in vitro models of EC cultures showed that the expression levels of ICAM-1 and VCAM-1 were significantly upregulated in the ECs treated with Gas6 (100-400 ng/mL; Figure 3A,D). Interestingly, our results showed that the treatment with 5 μmol/L UNC2250, a selective Mer tyrosine kinase inhibitor, suppressed the upregulation of ICAM-1 and VCAM-1, which was induced by Gas6 or GVHD sera in the ECs (Figure 3B-C,E-F). The sera of the patients without GVHD and with or without UNC2250 did not affect the ICAM-1 and VCAM-1 expression levels in their ECs (Figure 3C,F). Furthermore, 30-minute incubation with GVHD sera specifically induced the phosphorylation of Mer in the ECs (Figure 3Ga). These results suggest that Mer inhibition may have beneficial effects on endothelial dysfunction overlapping between GVHD by enhancing the adherence of inflammatory cells to ECs and the subsequent development of TA-TMA.
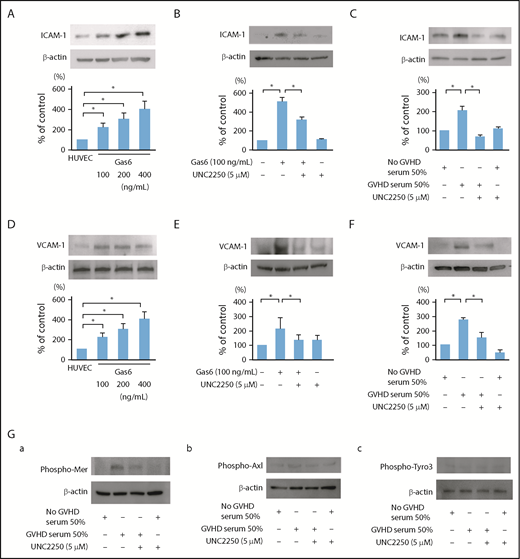
Effect of Mer inhibition on ICAM-1 and VCAM-1 upregulation induced by exogenous Gas6 or the exposure of sera isolated from patients with grade III aGVHD in ECs. (A,D) The expression of ICAM-1 and VCAM-1 in ECs was upregulated by exogenous Gas6. The ECs were incubated for 24 hours with exogenous Gas6 (0, 100, 200, 400 ng/mL), followed by western blotting. Representative data are from 4 independent experiments. *P < .05. (B,E) The expression levels of ICAM-1 and VCAM-1 were increased by exogenous Gas6, and 5 μmol/L UNC2250, a selective Mer tyrosine kinase inhibitor significantly suppressed ICAM-1 and VCAM-1 upregulation in the ECs, which were then incubated for 24 hours with exogenous Gas6 (100 ng/mL) with or without UNC2250 (5 μmol/L), followed by western blotting. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (C,F) The expression of ICAM-1 and VCAM-1 was significantly increased by the exposure of sera isolated from patients with grade III aGVHD, and 5 μmol/L UNC2250 suppressed the ICAM-1 and VCAM-1 upregulation in ECs. The ECs were incubated for 24 hours with the sera isolated from patients with or without grade III aGVHD at a level of 50 (vol/vol) of the total medium with or without UNC2250 (5 μmol/L), followed by western blotting. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (Ga-c) The effects of GVHD sera on the phosphorylation of Mer, Axl, and Tyro3 in the ECs. Thirty minutes of incubation of GVHD sera specifically induced the phosphorylation of Mer in the ECs. Representative data are from 3 independent experiments.
Effect of Mer inhibition on ICAM-1 and VCAM-1 upregulation induced by exogenous Gas6 or the exposure of sera isolated from patients with grade III aGVHD in ECs. (A,D) The expression of ICAM-1 and VCAM-1 in ECs was upregulated by exogenous Gas6. The ECs were incubated for 24 hours with exogenous Gas6 (0, 100, 200, 400 ng/mL), followed by western blotting. Representative data are from 4 independent experiments. *P < .05. (B,E) The expression levels of ICAM-1 and VCAM-1 were increased by exogenous Gas6, and 5 μmol/L UNC2250, a selective Mer tyrosine kinase inhibitor significantly suppressed ICAM-1 and VCAM-1 upregulation in the ECs, which were then incubated for 24 hours with exogenous Gas6 (100 ng/mL) with or without UNC2250 (5 μmol/L), followed by western blotting. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (C,F) The expression of ICAM-1 and VCAM-1 was significantly increased by the exposure of sera isolated from patients with grade III aGVHD, and 5 μmol/L UNC2250 suppressed the ICAM-1 and VCAM-1 upregulation in ECs. The ECs were incubated for 24 hours with the sera isolated from patients with or without grade III aGVHD at a level of 50 (vol/vol) of the total medium with or without UNC2250 (5 μmol/L), followed by western blotting. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (Ga-c) The effects of GVHD sera on the phosphorylation of Mer, Axl, and Tyro3 in the ECs. Thirty minutes of incubation of GVHD sera specifically induced the phosphorylation of Mer in the ECs. Representative data are from 3 independent experiments.
Effects of Mer inhibition on TM and PAI-1 expression induced by Gas6 or the exposure of sera isolated from patients with grade III aGVHD to ECs
The increased levels of soluble TM and PAI-1 correlated with HSCT-related vascular complications such as sinusoidal obstruction syndrome/TA-TMA and capillary leak syndrome, as well as with, to a lesser degree, GVHD.17,18,44 TM is an EC membrane glycoprotein that forms a complex with thrombin, converting thrombin from a procoagulant to an anticoagulant enzyme.18,44-46 PAI-1, which is secreted by ECs, is linked to endothelial dysfunction and coagulation activation during systemic inflammation.17,18,44 In the current study, Western immunoblots showed that overnight incubation with Gas6 (100-400 ng/mL) significantly induced the downregulation of TM and the upregulation of PAI-1 in ECs (Figure 4A,D). Crucially, Gas6 or GVHD sera induced the downregulation of TM and the upregulation of PAI-1 in the ECs, and Mer inhibition by UNC2250 inhibited the downregulation of TM and upregulation of PAI-1 in the ECs stimulated by Gas6 or GVHD sera (Figure 4B-C,E-F). The sera of the patients without aGVHD and those with UNC2250 did not affect the TM and PAI-1 expression levels in their ECs (Figure 4C,F). Autocrine and paracrine Gas6 enhances platelet degranulation and aggregation through TAM receptors, causing platelet αIIbβ3 activation and thrombus formation.47 Our real-time PCR analysis results showed that mRNA expressed Gas6 and Mer in the human platelets of the patients with or without aGVHD, and there was no significant difference between patients with and those without aGVHD (Figure 4Ga).In addition, Figure 4Gb showed that pretreatment with UNC2250 decreased the mRNA expression of Gas6 in the human platelets of the patients without aGVHD, and did not affect Mer mRNA expression. The platelet aggregation test showed that collagen or ADP with or without Gas6 increased platelet aggregation, which was markedly inhibited by UNC2250 (Figure 4H-K). These findings suggest that the Gas-Mer-related pathway may contribute to endothelial dysfunction, platelet aggregation, and coagulation activation in a common pathology between GVHD and TA-TMA in patients undergoing HSCT.

Gas6-Mer axis is involved in the expression of TM and PAI-1 in ECs. (A,D) The downregulation of TM and upregulation of PAI-1 were induced by exogenous Gas6. The ECs were incubated for 24 hours with exogenous Gas6 (0, 100, 200, 400 ng/mL), followed by western blotting. Representative data are from 4 independent experiments. *P < .05. (B,E) The downregulation of TM and upregulation of PAI-1 were increased by exogenous Gas6, and 5 μmol/L UNC2250, a selective Mer tyrosine kinase inhibitor, significantly inhibited the downregulation of TM and the upregulation of PAI-1 in the ECs, which were then incubated for 24 hours with exogenous Gas6 (100 ng/mL) with or without UNC2250 (5 μmol/L), followed by western blotting. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (C,F) The downregulation of TM and upregulation of PAI-1 were induced by the exposure of sera isolated from patients with grade III aGVHD at 50% (vol/vol) of the total media, and UNC2250 significantly inhibited the downregulation of TM and upregulation of PAI-1 in ECs. The ECs were incubated for 24 hours with the sera isolated from the patients with or without grade III aGVHD at a level of 50% (vol/vol) of the total medium with or without UNC2250 (5 μmol/L), followed by western blotting. Representative immunoblots from 4 similar experiments are shown. Bars are the means ± SD of quantitative densitometric analyses. *P < .05. (Ga-b) mRNA expressed Mer and Gas6 in human platelets of patients with or without aGVHD. To examine whether the platelet mRNA expression of Gas6 and Mer in human platelets is inhibited by UNC2250, real-time PCR was performed. UNC2250 (5 μmol/L) pretreatment (4 hours) decreased the mRNA expression of Gas6 in human platelets of patients without aGVHD, and did not affect the mRNA expression of Mer. Representative data from 3 experiments are shown. (H-K) The effect of UNC2250 on increased platelet aggregation induced by collagen (2.5 μg/mL) and ADP (5 μmol/L) with or without Gas6 (200 ng/mL). Representative data from 3 similar experiments are shown.
Gas6-Mer axis is involved in the expression of TM and PAI-1 in ECs. (A,D) The downregulation of TM and upregulation of PAI-1 were induced by exogenous Gas6. The ECs were incubated for 24 hours with exogenous Gas6 (0, 100, 200, 400 ng/mL), followed by western blotting. Representative data are from 4 independent experiments. *P < .05. (B,E) The downregulation of TM and upregulation of PAI-1 were increased by exogenous Gas6, and 5 μmol/L UNC2250, a selective Mer tyrosine kinase inhibitor, significantly inhibited the downregulation of TM and the upregulation of PAI-1 in the ECs, which were then incubated for 24 hours with exogenous Gas6 (100 ng/mL) with or without UNC2250 (5 μmol/L), followed by western blotting. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (C,F) The downregulation of TM and upregulation of PAI-1 were induced by the exposure of sera isolated from patients with grade III aGVHD at 50% (vol/vol) of the total media, and UNC2250 significantly inhibited the downregulation of TM and upregulation of PAI-1 in ECs. The ECs were incubated for 24 hours with the sera isolated from the patients with or without grade III aGVHD at a level of 50% (vol/vol) of the total medium with or without UNC2250 (5 μmol/L), followed by western blotting. Representative immunoblots from 4 similar experiments are shown. Bars are the means ± SD of quantitative densitometric analyses. *P < .05. (Ga-b) mRNA expressed Mer and Gas6 in human platelets of patients with or without aGVHD. To examine whether the platelet mRNA expression of Gas6 and Mer in human platelets is inhibited by UNC2250, real-time PCR was performed. UNC2250 (5 μmol/L) pretreatment (4 hours) decreased the mRNA expression of Gas6 in human platelets of patients without aGVHD, and did not affect the mRNA expression of Mer. Representative data from 3 experiments are shown. (H-K) The effect of UNC2250 on increased platelet aggregation induced by collagen (2.5 μg/mL) and ADP (5 μmol/L) with or without Gas6 (200 ng/mL). Representative data from 3 similar experiments are shown.
Mer inhibition suppressed EC apoptosis in vitro
EC apoptosis is the earliest target of an allogeneic GVH response, and the degree of EC apoptosis in the target organs of GVHD is associated with the severity of GVHD, leading to transmural microvascular lesions and capillary hemorrhage.14,48 As determined by TUNEL staining, in the present study, we showed that EC apoptosis was induced by GVHD sera isolated from 4 patients with grade III aGVHD at 25%, 50%, and 100% (vol/vol) of the total media (Figure 5A-C). UNC2250 significantly suppressed the EC apoptosis induced by GVHD sera at 50% (vol/vol) of the total media (Figure 5Dc-e). Pro-inflammatory cytokines such as TNF-α and IL-1β are reported to be associated with cell apoptosis after HSCT.5,7,49,50 In the current study, GVHD sera induced mRNA upregulation of TNF-α and IL-1β, which was inhibited by UNC2250 (Figure 5E-F), indicating that UNC2250 can potentially inhibit GVHD sera-induced endothelial apoptosis through the increase of TNF-α and IL-1β in ECs. Our results indicate that Mer may be responsible for the pathogenesis of EC apoptosis in HSCT-related complications.
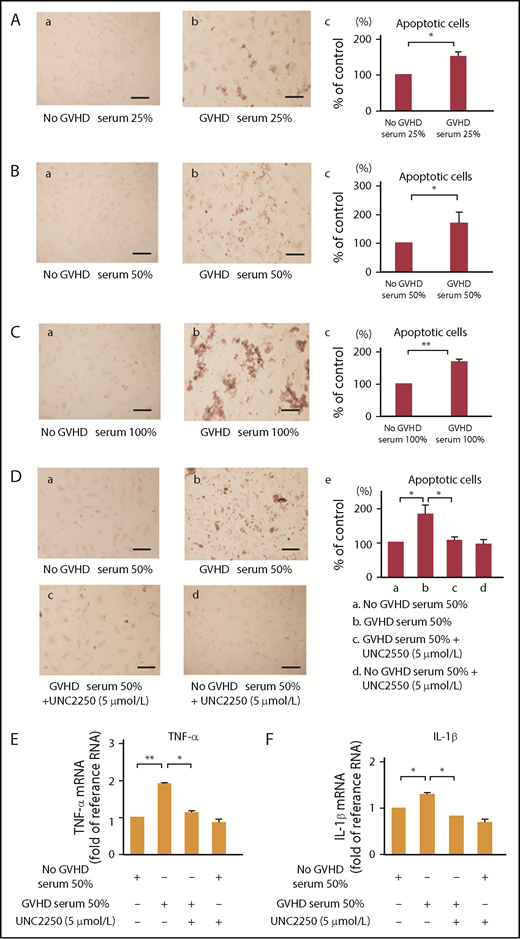
Mer inhibition inhibited endothelial cell apoptosis stimulated by GVHD sera. (A-C) To examine GVHD sera on the apoptosis of ECs, we performed TUNEL staining. ECs were incubated for 24 hours with the sera isolated from patients with or without grade III aGVHD at a level of 0%, 25%, 50%, and 100% (vol/vol) of the total medium, followed by TUNEL staining. Scale bars represent 20 µm. Representative data are from 3 independent experiments. (Ac,Bc,Cc) The statistical analysis indicated the effect of GVHD sera on the EC apoptosis. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05; **P < .01. (Da-c) UNC2250 (5 μmol/L) significantly suppressed the EC apoptosis induced by the exposure of sera isolated from patients with grade III aGVHD in ECs at 50% (vol/vol) of the total media. (De) The statistical analysis indicated the effect of exogenous Gas6 or the exposure of sera isolated from patients with grade III aGVHD to ECs on the EC apoptosis. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (E-F) GVHD sera induced mRNA upregulation of TNF-α and IL-1β, which was inhibited by UNC2250 in ECs. The ECs were incubated for 6 hours with the sera isolated from patients with or without grade III aGVHD at a level of 50% (vol/vol) of the total medium with or without 5 μmol/L UNC2250, followed by real-time PCR. Representative data from 4 similar experiments are shown. Bars are the means ± SD of quantitative densitometric analyses. **P < .01, *P < .05.
Mer inhibition inhibited endothelial cell apoptosis stimulated by GVHD sera. (A-C) To examine GVHD sera on the apoptosis of ECs, we performed TUNEL staining. ECs were incubated for 24 hours with the sera isolated from patients with or without grade III aGVHD at a level of 0%, 25%, 50%, and 100% (vol/vol) of the total medium, followed by TUNEL staining. Scale bars represent 20 µm. Representative data are from 3 independent experiments. (Ac,Bc,Cc) The statistical analysis indicated the effect of GVHD sera on the EC apoptosis. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05; **P < .01. (Da-c) UNC2250 (5 μmol/L) significantly suppressed the EC apoptosis induced by the exposure of sera isolated from patients with grade III aGVHD in ECs at 50% (vol/vol) of the total media. (De) The statistical analysis indicated the effect of exogenous Gas6 or the exposure of sera isolated from patients with grade III aGVHD to ECs on the EC apoptosis. Results are shown as mean ± SD of statistical analyses from 4 separate experiments. *P < .05. (E-F) GVHD sera induced mRNA upregulation of TNF-α and IL-1β, which was inhibited by UNC2250 in ECs. The ECs were incubated for 6 hours with the sera isolated from patients with or without grade III aGVHD at a level of 50% (vol/vol) of the total medium with or without 5 μmol/L UNC2250, followed by real-time PCR. Representative data from 4 similar experiments are shown. Bars are the means ± SD of quantitative densitometric analyses. **P < .01, *P < .05.
Preventive effects of Mer inhibition on GVHD and TA-TMA of liver and kidney in mouse HSCT models
Mouse HSCT models intravenously received 3 mg/kg UNC2250 at 14 days after HSCT, and were then sacrificed on day 21 after HSCT. As determined by flow cytometry with anti HLA-antibodies, we demonstrated successful mixed chimerism, which showed the expression of the donor’s H-2q haplotype in recipient bone marrow with intravenous administration of UNC2250 or not (Figure 6A). To assess the degree of GVHD, the body weight of the recipients was measured every other day, and the recipients were also assessed once per week on the clinical findings, using clinical GVHD scores.51 In mouse HSCT models, we observed rapid weight loss (GVHD scores, grade II), which was mitigated by UNC2250 (Figure 6B). The mouse HSCT models also had severe diarrhea, hair loss, skin lesions, or decreased motor activity, which were reduced by UNC2250 (data not shown). As shown in Figure 6C, the serum Gas6 levels were significantly increased, and UNC2250 then reduced the levels of Gas6 in the mouse HSCT models. Macroscopic findings showed that formation of multiple nodules, which were caused by inflammatory response in the progression of hepatic GVHD in mouse HSCT models, as well as UNC2250, inhibited the characteristic findings of hepatic GVHD in mouse HSCT models (Figure 6Fa-b). As determined by histological analyses, UNC2250 markedly inhibited the histological features of hepatic GVHD, showing hepatocellular necrosis, fibrosis, and apoptosis in mouse HSCT models (Figure 6G). TUNEL staining analysis also showed that apoptosis of liver cells was substantially reduced in the UNC2250-administered group compared with the non-UNC2250 group (Figure 6H). In addition, UNC2250, in an analysis of liver lysates samples by western blot and IHC (Figure 6D-E), was found to have downregulated the expression levels of Gas6 and Mer in the liver of HSCT mice. As determined by our histological analyses, formation of thrombi in hepatic and renal vessels was observed in the HSCT mice, and UNC2250 significantly suppressed the formation of thrombi in hepatic and renal vessels as compared with the groups without UNC2250 (Figure 6I-J). These findings indicate that Mer may be a therapeutic target for TA-TMA, which is identified by the formation of thrombi in the microvasculature after HSCT.
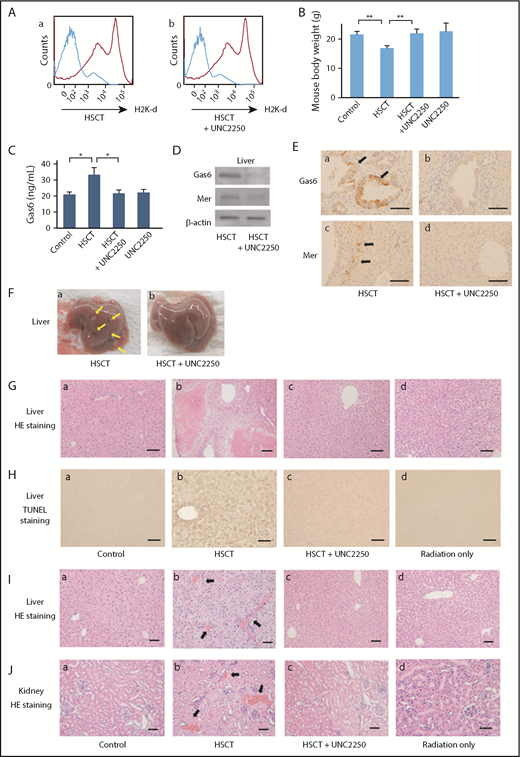
Effects of Mer inhibition on GVHD and TA-TMA of liver and kidney in mouse HSCT models Mouse HSCT models intravenously received 3 mg/kg UNC2250 at 14 days after HSCT, and the mouse models were sacrificed on day 21 after HSCT. (A) Mixed chimerism was determined by flow cytometry with anti HLA-antibodies, UNC2250 had no effect on engraftment in the HSCT mice. (B) The body weight of the recipient mice was assessed on the clinical findings using clinical GVHD scores. Body weight loss of the recipient mice was abrogated by intravenous administration of UNC2250. Data are expressed as mean ± SD (n = 9). **P < .01. (C) The levels of serum Gas6 were significantly increased in HSCT mice, using a mouse Gas6 ELISA kit. Data are expressed as mean ± SD (n = 9). *P < .05. (D) UNC2250 downregulated the expression of Gas6 and Mer in the livers of mouse HSCT models, using analysis of liver lysates samples by western blot. Representative data from 3 similar experiments are shown. (Ea-d) The intravenous administration of UNC2250 downregulated the expression of Gas6 and Mer in the mouse HSCT models. Immunohistochemical staining of Gas6 and Mer. Scale bars represent 5 µm. Original magnification ×200. (Fa-b) Macroscopic observations show that a formation of multiple nodules (arrows) was caused by inflammatory response in hepatic GVHD in mouse HSCT models. Intravenous administration of UNC2250 inhibited the characteristic findings of hepatic GVHD in the mouse HSCT models. (G) Hematoxylin and eosin (HE) stain. Scale bars represent 10 µm. Original magnification ×200. Histopathological findings showed that intravenous administration of UNC2250 markedly inhibited histological features of hepatic GVHD showing hepatocellular necrosis, fibrosis, and apoptosis in mouse HSCT models at 21 days after HSCT. (H) TUNEL staining analysis showed that apoptosis of liver cells was substantially reduced in the UNC2250-administered group compared with the non-UNC2250 group. Scale bars represent 5 µm. Original magnification ×200. (I-J) Histopathologic observations showed that formations of thrombi in hepatic and renal vessels were increased in mouse HSCT models at 21 days after HSCT, and intravenous administration of 3 mg/kg UNC2250 significantly suppressed the formations of thrombi in hepatic and renal vessels as compared with the group without treatment. Arrows indicate thrombi formation in vessels of the liver and kidney. Scale bars represent 10 µm. Original magnification ×200.
Effects of Mer inhibition on GVHD and TA-TMA of liver and kidney in mouse HSCT models Mouse HSCT models intravenously received 3 mg/kg UNC2250 at 14 days after HSCT, and the mouse models were sacrificed on day 21 after HSCT. (A) Mixed chimerism was determined by flow cytometry with anti HLA-antibodies, UNC2250 had no effect on engraftment in the HSCT mice. (B) The body weight of the recipient mice was assessed on the clinical findings using clinical GVHD scores. Body weight loss of the recipient mice was abrogated by intravenous administration of UNC2250. Data are expressed as mean ± SD (n = 9). **P < .01. (C) The levels of serum Gas6 were significantly increased in HSCT mice, using a mouse Gas6 ELISA kit. Data are expressed as mean ± SD (n = 9). *P < .05. (D) UNC2250 downregulated the expression of Gas6 and Mer in the livers of mouse HSCT models, using analysis of liver lysates samples by western blot. Representative data from 3 similar experiments are shown. (Ea-d) The intravenous administration of UNC2250 downregulated the expression of Gas6 and Mer in the mouse HSCT models. Immunohistochemical staining of Gas6 and Mer. Scale bars represent 5 µm. Original magnification ×200. (Fa-b) Macroscopic observations show that a formation of multiple nodules (arrows) was caused by inflammatory response in hepatic GVHD in mouse HSCT models. Intravenous administration of UNC2250 inhibited the characteristic findings of hepatic GVHD in the mouse HSCT models. (G) Hematoxylin and eosin (HE) stain. Scale bars represent 10 µm. Original magnification ×200. Histopathological findings showed that intravenous administration of UNC2250 markedly inhibited histological features of hepatic GVHD showing hepatocellular necrosis, fibrosis, and apoptosis in mouse HSCT models at 21 days after HSCT. (H) TUNEL staining analysis showed that apoptosis of liver cells was substantially reduced in the UNC2250-administered group compared with the non-UNC2250 group. Scale bars represent 5 µm. Original magnification ×200. (I-J) Histopathologic observations showed that formations of thrombi in hepatic and renal vessels were increased in mouse HSCT models at 21 days after HSCT, and intravenous administration of 3 mg/kg UNC2250 significantly suppressed the formations of thrombi in hepatic and renal vessels as compared with the group without treatment. Arrows indicate thrombi formation in vessels of the liver and kidney. Scale bars represent 10 µm. Original magnification ×200.
Discussion
In the present study, we demonstrated that the Gas6-Mer axis may contribute to the mechanisms underlying the progression of the pathogenesis of GVHD and TA-TMA. The signaling pathway may be responsible for endothelial dysfunction overlapping between GVHD and TA-TMA.3,19,40 We suggest that the Gas6-Mer axis can potentially be an attractive therapeutic target for treating GVHD and TA-TMA caused by systemic endothelial dysfunction after HSCT.
Gas6 is a ligand that binds to Tyro3, Axl, and Mer (TAM) receptors, which belong to a family of receptor tyrosine kinases.22,23 Gas6 has been known to promote inflammation by enhancing interactions among ECs, platelets, and leukocytes.22,30,52 It has been reported that there is an association between venous thromboembolism and elevated Gas6 levels consistent with in vivo murine models of thrombosis.53 Mer, which regulates platelet activation and thrombus formation, has been reported to be ectopically expressed in the majority of acute leukemias, and increasing evidence suggests a role for Mer in multiple solid tumors.25,26,54
In the present study, the serum Gas6 levels were significantly increased in HSCT patients 21 to 35 days after HSCT (Figure 1A). In addition, we found that the serum Gas6 levels were markedly increased in the HSCT patients with grade II to IV aGVHD compared with those with grade 0 to I aGVHD (Figure 1B). ES and capillary leak syndrome is caused by damage to the ECs, resulting in extravasation of plasma proteins and fluid from the capillaries into the extravascular space.37 Our ELISA analysis showed that the serum Gas6 levels were significantly increased in the patient group (n = 6) with ES compared with the patient group without ES (n = 8; Figure 1C). Endothelial dysfunction plays a role in posttransplant renal complications, and such dysfunction may not be reflected in the plasma levels of PAI-1 or d-dimer in the pathology of kidney injury resulting from TA-TMA and GVHD after HSCT.10,19,44 In the present study, LDH, resistance to platelet transfusion, creatinine levels, and thrombin-antithrombin complex values were significantly increased in patients with grade II to IV aGVHD compared with those with grade 0 to I aGVHD (Figure 1Fa-d), indicating a common pathology between GVHD and TA-TMA. The serum Gas6 levels were correlated with LDH, d-dimer, and plasmin inhibitor complex values in 3 representative cases with grade II to III aGVHD after HSCT (Figure 1G). Gas6 is expressed not only in many cell types, including blood cells, bone marrow cells, ECs, smooth muscle cells, and mesangial cells, but also in several cancer cells.22,52,54 In the present study, our flow cytometric analysis indicated that Gas6 was expressed in a distribution consistent with that in the CD3-positive lymphocytes and CD14-positive monocytes from the blood samples of the HSCT patients with aGVHD compared with the patients without aGVHD (Figure 1D-E). In the aGVHD lesions of the large intestine and skin, our findings showed that the expression of Gas6 and Mer receptor was significantly upregulated (Figure 2Bc-f,Ca-b). Double IHC staining showed that Gas6 bound to Mer in aGVHD lesions of the large intestine and skin (Figure 2Bi-k,Ce-g), Our data indicate that the Gas6-Mer axis can potentially play key roles in the pathology of GVHD.
EC damage occurring in early-phase HSCT plays critical roles in all events during HSCT-associated complications, including GVHD and TA-TMA.3,19,40 The elevations of soluble ICAM-1 and VCAM-1 levels precede the appearance of clinical symptoms in patients with TA-TMA and/or GVHD,16,55,56 and the adherence of activated T cells and leukocytes to ECs mediated via ICAM-1 and VCAM-1 subsequently induces GVHD progression16,55-57 In the present study, Gas6 or GVHD sera induced the upregulation of ICAM-1 and VCAM-1, which was reversed by UNC2250 in the ECs (Figures 3B-C,E-F). Therefore, these findings suggest that the Gas6-Mer axis may be involved in a causal link between GVHD and the subsequent development of TA-TMA, by enhancing the adherence of inflammatory cells to the ECs. TM and PAI-1 were identified as biomarkers of EC dysfunction/activation after HSCT, and the increases of TM and PAI-1 have been associated with patients developing TA-TMA as well as GVHD.17,18,44,58 UNC2250 significantly suppressed the downregulation of TM and the upregulation of PAI-1 in the ECs stimulated by Gas6 or GVHD sera (Figure 4B-C,E-F). The platelet aggregation test showed that collagen (2.5 μg/mL) or ADP (5 μmol/L) with or without Gas6 induced increased platelet aggregation, which was markedly inhibited by Mer inhibition by UNC2250 (Figure 4H-K). Mer inhibition leading to reduced platelet aggregation may be a risk factor for bleeding in HSCT patients experiencing long-term thrombocytopenia. However, a previous study reported that Mer inhibition protected murine models from pulmonary embolism and arterial thrombosis without increasing bleeding times, and that spontaneous hemorrhage or bleeding time did not increase after tail clipping in Mer-deficient mice.30,59 At the concentration of 3 mg/kg UNC2250 we used for our in vivo experiments, no bleeding signs were seen in the HSCT mice. Our findings suggest that Mer may be responsible for the overlapping mechanisms of the hypercoagulable status associated with EC dysfunction, underlying the pathogenesis in TA-TMA and GVHD.
EC apoptosis induced by pro-inflammatory cytokines could be an important mechanism of vascular injury, resulting in vascular leak, inflammation, and coagulation.5,7,50,60,61 In the present study, EC apoptosis was induced by GVHD sera at 25%, 50%, and 100% (vol/vol) of the total media (Figure 5A-C). Inhibition of Mer suppressed the EC apoptosis induced by GVHD sera (Figure 5Da-d). In addition, GVHD sera induced mRNA upregulation of TNF-α and IL-1β, which was inhibited by UNC2250 (Figure 5E-F). These results indicate that Mer inhibition by UNC2250 may inhibit endothelial apoptosis through increases in TNF-α and IL-1β in the ECs stimulated by GVHD sera. However, further studies are required to confirm these findings. We suggest that the Gas6-Mer-related signaling pathway may be a critical modifier for EC apoptosis involved in aGVHD.
In our mouse HSCT models, the serum Gas6 levels were significantly increased, and UNC2250 then reduced the Gas6 levels in the HSCT mice (Figure 6C). UNC2250 has been reported to be specific for Mer, but also targets other pathways such as serine/threonine kinases.38 This suggests that UNC2250 may have inhibited the increased serum Gas6 levels in our HSCT mice in addition to blocking pro-inflammatory cytokines such as TNF-α and IL-1β via other pathways. To further address the effects of Mer inhibition on the pathology of GVHD and TA-TMA, we performed in vivo experiments in mouse HSCT models. Surprisingly, as determined by macroscopic and histological analyses, the UNC2250 markedly attenuated the histopathological characteristics of GVHD and TA-TMA of the liver and kidney in mouse HSCT models (Figure 6G-I,J). TUNEL staining analysis showed that UNC2250 markedly reduced hepatocyte apoptosis in our mouse models (Figure 6H). In addition, we demonstrated that UNC2250 inhibited vascular thrombus formation in the liver and kidney (Figure 6I-J). Interestingly, UNC2250 reduced the expression of Gas6 and Mer in the liver of the HSCT mice (Figures 6D-E), indicating that UNC2250 may lead to Mer downregulation as well as reduced Mer activity. However, further research is needed to fully understand the underlying mechanisms.
In conclusion, the present study offers new insights into the beneficial effects of Gas-Mer inhibition on endothelial dysfunction contributing to the common pathology between GVHD and TA-TMA at an early phase after HSCT (Figure 7). On the basis of our findings, we suggest that the Gas6-Mer axis can potentially be a major modulator and may be a novel therapeutic target for the treatment of endothelial dysfunction, contributing to the overlapping pathology between GVHD and TA-TMA.
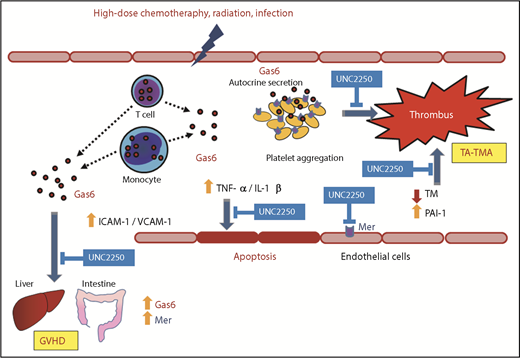
Schematic diagram describing the effects of the Gas6-Mer axis on endothelial dysfunction in the overlapping pathology between GVHD and TA-TMA Gas6 may be secreted from donor lymphocytes and monocytes in the peripheral blood of patients with GVHD. Gas6 mediated via Mer tyrosine kinase receptor induced platelet aggregation and hypercoagulable status, leading to subsequent TA-TMA onset. In addition, Gas6-Mer signaling pathway enhances progressive endothelial impairment in the common pathology between GVHD and TA-TMA.
Schematic diagram describing the effects of the Gas6-Mer axis on endothelial dysfunction in the overlapping pathology between GVHD and TA-TMA Gas6 may be secreted from donor lymphocytes and monocytes in the peripheral blood of patients with GVHD. Gas6 mediated via Mer tyrosine kinase receptor induced platelet aggregation and hypercoagulable status, leading to subsequent TA-TMA onset. In addition, Gas6-Mer signaling pathway enhances progressive endothelial impairment in the common pathology between GVHD and TA-TMA.
Acknowledgments
The authors acknowledge the outstanding technical assistance of Ayumi Haneda and Chisato Kubo.
This work was supported by the Fukushima Medical University Research Project and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (17K09959 and 18K08366). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authorship
Contribution: M. Furukawa, X.W., and H.O. designed the experiments, analyzed the data, and wrote the manuscript; M. Furukawa, X.W., H.O. M. Fukatsu, L.A., H.T., K.H.-S., A.S.-N., and S.K. performed the experiments; K.O. contributed to the research design; and T.I. interpreted the data, wrote the manuscript, and gave the final approval.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hiroshi Ohkawara, Department of Hematology, Fukushima Medical University, 1 Hikarigaoka, Fukushima 960-1295, Japan; e-mail: ohkawara@fmu.ac.jp.

