

Key Points
Polymorphisms of the FCGR2/3 locus are associated with susceptibility to childhood ITP and progression to chronic disease.
Genotyping of the FCGR2/3 locus may be helpful to determine prognosis and personalize treatment decisions in childhood ITP.

Abstract
In childhood immune thrombocytopenia (ITP), anti-platelet autoantibodies mediate platelet clearance through Fc-γ receptor (FcγR)–bearing phagocytes. In 75% to 90% of patients, the disease has a transient, self-limiting character. Here we characterized how polymorphisms of FcγR genes affect disease susceptibility, response to intravenous immunoglobulin (IVIg) treatment, and long-term recovery from childhood ITP. Genotyping of the FCGR2/3 locus was performed in 180 children with newly diagnosed ITP, 22 children with chronic ITP, and 180 healthy control children by multiplex ligation-dependent probe amplification. Children with newly diagnosed ITP were randomly assigned to a single administration of IVIg or observation, and followed for 1 year (Treatment With or Without IVIg for Kids With ITP [TIKI] trial). We defined transient ITP as a complete recovery (≥100 × 109/L) 3 months after diagnosis, including both self-limiting disease/IVIg responders and chronic ITP as absence of a complete recovery at 12 months. ITP susceptibility, as well as spontaneous recovery and response to IVIg, was associated with the genetic variants FCGR2C*ORF and FCGR2A*27W and the FCGR2B promoter variant 2B.4. These variants were overrepresented in patients with transient (N = 131), but not chronic (N = 43), disease. The presence of FCGR2C*ORF predisposed to transient ITP with an odds ratio of 4.7 (95% confidence interval, 1.9-14.3). Chronic ITP was associated with a deletion of FCGR2C/FCGR3B (copy number region 1) with an odds ratio of 6.2 (95% confidence interval, 1.8-24.7). Taken together, susceptibility to transient and chronic ITP is distinctly affected by polymorphic variants of FCGR2/3 genes. Our data suggest that genotyping of the FCGR2/3 locus may be useful for prognosis and guidance of treatment decisions in newly diagnosed childhood ITP.
Introduction
Childhood immune thrombocytopenia (ITP) is an acquired autoimmune bleeding disorder with an incidence of 1.9 to 6.4 per 100 000 children annually.1 Many children develop ITP after a mild viral infection.2-4 The disease is self-limiting in a large proportion of patients, and 75% to 90% of children will recover spontaneously within 6 to 12 months.2-6 Treatment with intravenous immunoglobulin (IVIg) shortens thrombocytopenia and prevented bleeding symptoms, but does not prevent chronic ITP.6 In particular, patients who do not respond to IVIg show an increased rate of chronic ITP.6,7 To date, it remains unresolved why some children develop chronic disease, and no specific biomarkers are available to determine prognosis.
Although the pathogenesis of ITP is complex, many patients have anti-platelet glycoprotein-specific autoantibodies, leading to accelerated clearance of opsonized platelets by Fc-γ receptor (FcγR)-bearing phagocytes, particularly in the spleen.8-10 In humans, FcγRs can be divided into activating (FcγRI, FcγRIIa, FcγRIIc, FcγRIIIa, FcγRIIIb) and inhibitory (FcγRIIb) FcγRs, based on intracellular signaling motifs. FcγR-encoding genes are subject to single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) that affect FcγR expression and function.11 Within ITP, FcγR can have an effect on multiple levels, such as on antigen presentation, B-cell activation threshold, a direct effect on platelet clearance through FcγR on myeloid and/or natural killer (NK) cells, and response to IVIg therapy.11-13 Previous studies have shown that variants of the FCGR2/3 locus are genetic risk factors for ITP, including the FCGR2A*131H and FCGR3A*158V alleles and the FCGR2C*ORF variant.14-20 As a result of the extensive linkage disequilibrium at the FCGR2/3 locus, it is not yet clear whether the reported variants are functionally associated with the disease. More important, the association of these genetic variants with disease courses (ie, response to IVIg, spontaneous recovery, and development of chronic disease) remains unresolved.
In the present study, we used longitudinal data for a cohort of children with newly diagnosed ITP who were followed for 1 year after initial diagnosis, as well as a second cross-sectional cohort of children with chronic ITP, to evaluate the association of all known FCGR2/3 polymorphisms with disease susceptibility, patient prognosis, and treatment responses to IVIg in childhood ITP.
Methods
Study participants
Children with newly diagnosed ITP, aged from 3 months to 16 years, a platelet count of 20 × 109/L or less, and with mild to moderate bleeding (grade 1-3 on the adapted Buchanan bleeding score21 ) were eligible for inclusion in the Treatment With or Without IVIg for Kids With ITP (TIKI) study.6 Patients were excluded if they had severe bleeding at diagnosis (Buchanan score >3), received immunomodulating drugs within 1 month before diagnosis, or suffered from conditions with a contraindication for IVIg, or if comprehension of the Dutch language was insufficient to give informed consent. Patients were randomly assigned to receive either a single-dose 0.8 g/kg bodyweight IVIg (Nanogam, Sanquin, The Netherlands) or to receive careful observation. Full blood counts were performed at diagnosis and during follow-up at 1 week, 1 month, 3 months, 6 months, and 12 months. DNA was isolated from samples obtained at diagnosis that were available for 180 of 200 patients. Response to IVIg was defined according to international guidelines.22 Parents and patients aged 12 years and older gave written informed consent. The study was registered in the Dutch Trial register (www.trialregister.nl; study ID 1563), approved by the Institutional Review Board of University Medical Center Utrecht, and performed in accordance with the Declaration of Helsinki.
Children with established chronic ITP were recruited for participation in a cross-sectional multicenter cohort study at outpatient clinics in the Chronic ITP in the Netherlands in Kids (CINKID) study.23 Children aged from 6 months to 17 years with chronic ITP were eligible. Exclusion criteria were presence of other autoimmune phenomena, presence of cytopenias besides thrombocytopenia (eg, hemoglobin <9.67 g/dL, leukocytes <4 × 109/L), or clinical or laboratory features suggestive of hereditary thrombocytopenia. Parents and patients aged 12 years and older gave written informed consent. The study was approved by the Institutional Review Board of University Medical Center Utrecht and performed in accordance with the Declaration of Helsinki.
Control samples were obtained from healthy volunteers participating in our institutional blood donor system (Sanquin, Amsterdam, The Netherlands).
DNA isolation and multiplex ligation-dependent probe amplification
Genomic DNA was isolated from whole blood, with a DNA extraction kit (QIAamp DNA blood mini kit, Qiagen Benelux, Venlo, The Netherlands). Multiplex ligation-dependent probe amplification, specifically designed for determination of genetic variations within the FCGR2/3 locus, was performed as described previously17,24-26 according to the manufacturers’ instructions (MRC Holland, Amsterdam, The Netherlands). This multiplex ligation-dependent probe amplification identifies SNPs and CNVs in the low-affinity FcγR genes; namely, FCGR2A (encoding for FcγRIIa with 2 possible allelic variants, 131H/R and 27Q/W), FCGR2B (232I/T), FCGR2C (Stop/ORF/nc-ORF), FCGR3A (158V/F), and FCGR3B (NA1/NA2/SH). In addition, the FCCR2B and FCGR2C promoter variants (−386G/C and −120A/T) were determined. The procedure has been described in detail previously.17 Data were analyzed with Genemarker, version 2.6.3. (Soft Genetics, State College, PA) and assessed in relation to 3 reference samples representing all known allotypic variants with predetermined CNVs. Haplotypes were determined as previously described.27 In brief, FCGR2C*ORF was scored in case of the presence of an exon 3 open reading frame (c.169C; rs759550223) and absence of the intron 7 splice variant (c.798+1A; rs76277413). In the case of c.169C and c.798+1A, the haplotype was scored as FCGR2C*nc-ORF (nonclassic open reading frame). When more alleles of c.169C were present than of c.798+1A, FCGR2C*ORF and FCGR2C*nc-ORF were scored as present. The FCGR2B/FCGR2C promoter variant 2B.4 was scored for any allele −386C (rs143796418; rs149754834) combined with allele −120A (rs780467580; rs34701572).
Ethnicity analyses
The prevalence of FCGR2/3 locus polymorphisms are significantly skewed between various ethnic groups.28,29 Ethnicity was determined by analysis of 15 autosomal short tandem repeat loci, using the PowerPlex 16 System (Promega, Madison, WI). Caucasian ethnicity was determined when the likelihood of support for Caucasian ethnicity exceeded 2 times the likelihood of support for Asian and African descent. Inclusion of healthy control children and patients with chronic childhood ITP (CINKID) was restricted to Caucasians because of, respectively, excess availability and the inability to compare ethnic differences in genotype frequencies in a small cohort. Nine of 31 CINKID patients from the original cohort were excluded, and 22 patients were left for analysis.
Meta-analysis
Studies were identified by a systematic search of EMBASE and PubMed (supplemental Table 1). Study characteristics are reported in supplemental Table 2. Meta-analyses were performed for each variant separately in R.30 Mantel-Haenszel odds ratios were estimated using fixed effects models.
Statistics
Statistical analysis was performed with R version 3.5.1 (July 2018; R Core Team). Genotype frequencies were analyzed by Fisher’s exact test. Effect sizes (odds ratios) were estimated by binomial logistic regression. Multiple testing correction was performed with false discovery rate, which is an overly conservative estimation when used on SNPs that are in linkage disequilibrium. Haplotype frequencies and posterior probabilities for each observation were estimated using the haplo.stats package.31 Kaplan-Meier curves were constructed with the survival package and tested with a log-rank test with a fixed covariate for group assignment.32 P < .05 was considered to be statistically significant. The sample size was determined by availability from existing cohorts.
Results
The present study included data from 180 children and adolescents with newly diagnosed ITP (TIKI cohort) and 22 patients with chronic ITP (CINKID cohort) from 2 separate multicenter cohort studies in the Netherlands (Table 1). In addition, we included 180 healthy control participants. At diagnosis, patients with ITP in both cohorts had a median age of 4 years, and approximately 55% experienced mucocutaneous bleeding symptoms. Patients in the CINKID study had a median disease duration of 5.4 years (interquartile range, 2.3-7.0 years).
Baseline characteristics
| Healthy controls (N = 180) | Newly diagnosed ITP (TIKI) (N = 180) | Chronic ITP (CINKID) (N = 22) | |
|---|---|---|---|
| Female, n/N (%) | 133/180 (74) | 84/180 (47) | 14/22 (64) |
| Caucasian, n/N (%) | 180/180 (100) | 123/135 (91) | 22/22 (100) |
| Age at diagnosis, y | 4.1 (2.6 - 7.7) | 3.8 (2.1-9.6) | |
| Preceding infection, n/N (%) | 96/178 (54) | 9/16 (56) | |
| Buchanan ccore >2, n/N (%) | 72/179 (40) | 7/22 (32) | |
| Presenting platelet count, ×109/L | 6 (3 - 10) | 15 (10-34) |
| Healthy controls (N = 180) | Newly diagnosed ITP (TIKI) (N = 180) | Chronic ITP (CINKID) (N = 22) | |
|---|---|---|---|
| Female, n/N (%) | 133/180 (74) | 84/180 (47) | 14/22 (64) |
| Caucasian, n/N (%) | 180/180 (100) | 123/135 (91) | 22/22 (100) |
| Age at diagnosis, y | 4.1 (2.6 - 7.7) | 3.8 (2.1-9.6) | |
| Preceding infection, n/N (%) | 96/178 (54) | 9/16 (56) | |
| Buchanan ccore >2, n/N (%) | 72/179 (40) | 7/22 (32) | |
| Presenting platelet count, ×109/L | 6 (3 - 10) | 15 (10-34) |
Data are median (interquartile range) unless noted otherwise.
Association of FCGR2/3 polymorphisms with susceptibility to childhood ITP
We first evaluated the association between FCGR2/3 CNV and polymorphisms with susceptibility to childhood ITP. Europeans show a strong linkage disequilibrium among FCGR2A*27W, FCGR2C*ORF, FCGR3A*158V, and the 2B.4 promoter.29,33 The variant FCGR2C*ORF was overrepresented in ITP (Table 2), with an odds ratio for ITP of 1.84 (95% confidence interval [CI], 1.14-2.98). In addition, a higher frequency of the FCGR2A*27W allele was observed in patients with ITP, indicating an association with ITP susceptibility (Table 2). In comparison with FCGR2A*27Q/Q, the 27Q/W genotype odds ratio for ITP was 1.94 (95% CI, 1.17-3.29). The FCGR2A*27W/W variant was present in only 4 individuals with ITP and 5 control individuals. The odds ratio of the linked FCGR2B/FCGR2C promoter 2B.4 for ITP was 1.42 (95% CI, 0.87-2.35). The FCGR3A*158V allele, which has been identified by multiple studies as a ITP susceptibility variant, was also enriched in our population, but the frequencies were not statistically different (Table 2). None of the genotypes associated with ITP remained significant when multiple testing corrections were performed. We performed meta-analyses of the identified loci, combining our present and previous studies, and ascertained the association of FCGR2C*ORF and FCGR3A*158V/F with susceptibility to childhood ITP (Figure 1).
Susceptibility to childhood immune thrombocytopenia
| Healthy controls (N = 180) | Newly diagnosed ITP (N = 180) | P | P (FDR adjusted) | OR (95% CI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | ||||
| Number of copies | |||||||||||||
| CNR1 | 0 (0) | 12 (6) | 155 (86) | 12 (7) | 1 (1) | 0 (0) | 12 (7) | 152 (84) | 16 (9) | 0 (0) | .78 | 1.0 | |
| CNR2 | 0 (0) | 1 (1) | 168 (93) | 11 (6) | 0 (0) | 0 (0) | 0 (0) | 173 (96) | 7 (4) | 0 (0) | .35 | 1.0 | |
| CNR3 | 0 (0) | 0 (0) | 180 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 180 (100) | 0 (0) | 0 (0) | ND | ND | |
| Alleles | |||||||||||||
| FCGR2A*27W | 146 (81) | 29 (16) | 5 (3) | 0 (0) | 0 (0) | 127 (71) | 49 (27) | 4 (2) | 0 (0) | 0 (0) | .033 | 0.30 | 1.94 (1.17-3.29)* |
| FCGR2A*131H | 38 (21) | 87 (48) | 55 (31) | 0 (0) | 0 (0) | 39 (22) | 95 (53) | 46 (26) | 0 (0) | 0 (0) | .56 | 1.0 | |
| FCGR2B*232T | 143 (79) | 31 (17) | 6 (3) | 0 (0) | 0 (0) | 137 (76) | 40 (22) | 3 (2) | 0 (0) | 0 (0) | .34 | 1.0 | |
| FCGR2C*ORF | 143 (79) | 32 (18) | 5 (3) | 0 (0) | 0 (0) | 122 (68) | 56 (31) | 2 (1) | 0 (0) | 0 (0) | .007 | .08 | 1.84 (1.14-2.98)† |
| FCGR2C*ncORF | 173 (96) | 3 (2) | 4 (2) | 0 (0) | 0 (0) | 170 (94) | 3 (2) | 7 (4) | 0 (0) | 0 (0) | .64 | 1.0 | |
| FCGR3A*158V | 79 (44) | 78 (43) | 23 (13) | 0 (0) | 0 (0) | 62 (34) | 85 (47) | 33 (18) | 0 (0) | 0 (0) | .14 | 1.0 | |
| FCGR3B*NA2 | 31 (17) | 79 (44) | 68 (38) | 2 (1) | 0 (0) | 20 (11) | 83 (46) | 77 (43) | 0 (0) | 0 (0) | .17 | 1.0 | |
| FCGR3B*SH | 173 (96) | 7 (4) | 0 (0) | 0 (0) | 0 (0) | 172 (96) | 8 (4) | 0 (0) | 0 (0) | 0 (0) | 1.0 | 1.0 | |
| FCGR2 promoter 2B.4 | 145 (81) | 31 (17) | 4 (2) | 0 (0) | 0 (0) | 134 (74) | 46 (26) | 0 (0) | 0 (0) | 0 (0) | .020 | .20 | 1.42 (0.87-2.35)† |
| Healthy controls (N = 180) | Newly diagnosed ITP (N = 180) | P | P (FDR adjusted) | OR (95% CI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | ||||
| Number of copies | |||||||||||||
| CNR1 | 0 (0) | 12 (6) | 155 (86) | 12 (7) | 1 (1) | 0 (0) | 12 (7) | 152 (84) | 16 (9) | 0 (0) | .78 | 1.0 | |
| CNR2 | 0 (0) | 1 (1) | 168 (93) | 11 (6) | 0 (0) | 0 (0) | 0 (0) | 173 (96) | 7 (4) | 0 (0) | .35 | 1.0 | |
| CNR3 | 0 (0) | 0 (0) | 180 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 180 (100) | 0 (0) | 0 (0) | ND | ND | |
| Alleles | |||||||||||||
| FCGR2A*27W | 146 (81) | 29 (16) | 5 (3) | 0 (0) | 0 (0) | 127 (71) | 49 (27) | 4 (2) | 0 (0) | 0 (0) | .033 | 0.30 | 1.94 (1.17-3.29)* |
| FCGR2A*131H | 38 (21) | 87 (48) | 55 (31) | 0 (0) | 0 (0) | 39 (22) | 95 (53) | 46 (26) | 0 (0) | 0 (0) | .56 | 1.0 | |
| FCGR2B*232T | 143 (79) | 31 (17) | 6 (3) | 0 (0) | 0 (0) | 137 (76) | 40 (22) | 3 (2) | 0 (0) | 0 (0) | .34 | 1.0 | |
| FCGR2C*ORF | 143 (79) | 32 (18) | 5 (3) | 0 (0) | 0 (0) | 122 (68) | 56 (31) | 2 (1) | 0 (0) | 0 (0) | .007 | .08 | 1.84 (1.14-2.98)† |
| FCGR2C*ncORF | 173 (96) | 3 (2) | 4 (2) | 0 (0) | 0 (0) | 170 (94) | 3 (2) | 7 (4) | 0 (0) | 0 (0) | .64 | 1.0 | |
| FCGR3A*158V | 79 (44) | 78 (43) | 23 (13) | 0 (0) | 0 (0) | 62 (34) | 85 (47) | 33 (18) | 0 (0) | 0 (0) | .14 | 1.0 | |
| FCGR3B*NA2 | 31 (17) | 79 (44) | 68 (38) | 2 (1) | 0 (0) | 20 (11) | 83 (46) | 77 (43) | 0 (0) | 0 (0) | .17 | 1.0 | |
| FCGR3B*SH | 173 (96) | 7 (4) | 0 (0) | 0 (0) | 0 (0) | 172 (96) | 8 (4) | 0 (0) | 0 (0) | 0 (0) | 1.0 | 1.0 | |
| FCGR2 promoter 2B.4 | 145 (81) | 31 (17) | 4 (2) | 0 (0) | 0 (0) | 134 (74) | 46 (26) | 0 (0) | 0 (0) | 0 (0) | .020 | .20 | 1.42 (0.87-2.35)† |
Data are n (% of cohort). Number of copies are given for the respective copy number region (CNR). Frequency P value by Fisher's exact test. Bold values denote statistically significant P values. FCGR3B*NA2 and FCGR3B*SH are also described as FCGR3B*02 and FCGR3B*03 according to recent nomenclature.51
CI, confidence interval; FDR, adjustment of P values by false discovery rate; ND, not performed; OR, odds ratio.
OR given for comparison of 27Q/W vs 27Q/W in association with ITP.
OR given for presence of 1 or 2 copies vs absence of the variant in association with ITP.
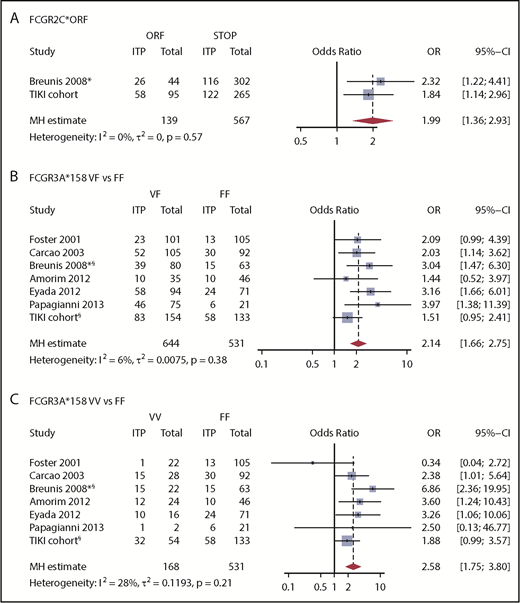
Meta-analysis for the association of FCGR2/3 variants with susceptibility to childhood ITP. Previous studies analyzing the association of FCGR2/3 with childhood ITP were identified in PubMed and EMBASE by systematic literature search (supplemental Table 1). The study by Bruin et al16 was not included because these patients were also included by Breunis et al.17 Study characteristics are reported in supplemental Table 2. (A) FCGR2C*ORF is significantly associated with susceptibility to childhood ITP. In comparison with FCGR3A*158F/F, FCGR3A*158V/F (B) and FCGR3A*158V/V (C) confer susceptibility to childhood ITP. This genetic variant is in linkage disequilibrium with FCGR2C*ORF, FCGR2A*27Q/W, and the FCGR2B/FCGR2C promoter polymorphism 2B.4. Mantel-Haenszel (MH) estimates of the ORs are given for fixed effect models. *Data were only analyzed from childhood ITP cases included in this study. §Patients with CNV in FCGR3A were excluded, whereas other studies did not supply such data.
Meta-analysis for the association of FCGR2/3 variants with susceptibility to childhood ITP. Previous studies analyzing the association of FCGR2/3 with childhood ITP were identified in PubMed and EMBASE by systematic literature search (supplemental Table 1). The study by Bruin et al16 was not included because these patients were also included by Breunis et al.17 Study characteristics are reported in supplemental Table 2. (A) FCGR2C*ORF is significantly associated with susceptibility to childhood ITP. In comparison with FCGR3A*158F/F, FCGR3A*158V/F (B) and FCGR3A*158V/V (C) confer susceptibility to childhood ITP. This genetic variant is in linkage disequilibrium with FCGR2C*ORF, FCGR2A*27Q/W, and the FCGR2B/FCGR2C promoter polymorphism 2B.4. Mantel-Haenszel (MH) estimates of the ORs are given for fixed effect models. *Data were only analyzed from childhood ITP cases included in this study. §Patients with CNV in FCGR3A were excluded, whereas other studies did not supply such data.
By estimating haplotype frequencies from the observed data, we confirmed a linkage disequilibrium in the present study (Table 3), but the low sample size did not allow us to directly associate them with ITP susceptibility.
Linkage disequilibrium among FCGR2A*27W, FCGR2C*ORF, 2B.4, and FCGR3A*158V determines major observed haplotypes
| FCGR2A-27W | FCGR2C-ORF, rs759550223 | FCGR2C-ncORF, rs76277413 | FCGR2B-2B.4, rs148754834; rs34701572 | FCGR3A-158V | Haplotype frequency | |
|---|---|---|---|---|---|---|
| Controls | Cases | |||||
| Q | STOP | WT | no 2B.4 | F | 0.6390 | 0.5580 |
| Q | STOP | WT | no 2B.4 | V | 0.1880 | 0.2060 |
| Q | STOP | WT | 2B.4 | F | 0.0319 | 0.0150 |
| Q | STOP | WT | 2B.4 | V | NA | 2.2e-07 |
| Q | ORF | WT | no 2B.4 | F | 5.9e-10 | 0.0055 |
| Q | ORF | splice* | no 2B.4 | V | 0.0103 | 0.0172 |
| Q | ORF | WT | no 2B.4 | V | 0.0036 | 0.0016 |
| Q | ORF | WT | 2B.4 | F | 3.5e-09 | NA |
| Q | ORF | WT | 2B.4 | V | 0.0102 | 0.0174 |
| W | STOP | WT | no 2B.4 | F | 0.0036 | 0.0015 |
| W | STOP | WT | no 2B.4 | V | 0.0036 | 0.0102 |
| W | STOP | WT | 2B.4 | F | 0.0034 | 0.0056 |
| W | STOP | WT | 2B.4 | V | NA | 0.0036 |
| W | ORF | WT | no 2B.4 | F | NA | 0.0042 |
| W | ORF | WT | no 2B.4 | V | 0.0317 | 0.0544 |
| W | ORF | WT | 2B.4 | V | 0.0743 | 0.0997 |
| FCGR2A-27W | FCGR2C-ORF, rs759550223 | FCGR2C-ncORF, rs76277413 | FCGR2B-2B.4, rs148754834; rs34701572 | FCGR3A-158V | Haplotype frequency | |
|---|---|---|---|---|---|---|
| Controls | Cases | |||||
| Q | STOP | WT | no 2B.4 | F | 0.6390 | 0.5580 |
| Q | STOP | WT | no 2B.4 | V | 0.1880 | 0.2060 |
| Q | STOP | WT | 2B.4 | F | 0.0319 | 0.0150 |
| Q | STOP | WT | 2B.4 | V | NA | 2.2e-07 |
| Q | ORF | WT | no 2B.4 | F | 5.9e-10 | 0.0055 |
| Q | ORF | splice* | no 2B.4 | V | 0.0103 | 0.0172 |
| Q | ORF | WT | no 2B.4 | V | 0.0036 | 0.0016 |
| Q | ORF | WT | 2B.4 | F | 3.5e-09 | NA |
| Q | ORF | WT | 2B.4 | V | 0.0102 | 0.0174 |
| W | STOP | WT | no 2B.4 | F | 0.0036 | 0.0015 |
| W | STOP | WT | no 2B.4 | V | 0.0036 | 0.0102 |
| W | STOP | WT | 2B.4 | F | 0.0034 | 0.0056 |
| W | STOP | WT | 2B.4 | V | NA | 0.0036 |
| W | ORF | WT | no 2B.4 | F | NA | 0.0042 |
| W | ORF | WT | no 2B.4 | V | 0.0317 | 0.0544 |
| W | ORF | WT | 2B.4 | V | 0.0743 | 0.0997 |
Data shown for 146 control individuals and 145 patients with newly diagnosed ITP (TIKI) who showed no CNV in CNR1 or CNR2. Bold text indicates the most common haplotypes. Frequencies of haplotypes are derived from posterior probabilities using observed alleles using the haplo.stats package in R. 2B.4 promoter constitutes the observation of a A at position −120 and C at position −386. The 2B.4 haplotype is solely observed as a promoter polymorphism of FCGR2B and SNPs are only indicated for this variant.
NA, haplotype not observed.
CNV is present in 4 distinct regions (CNRs) encompassing multiple genes (Figure 2A).24,34 In accordance with previous studies, there was no skewing in CNV at the FcγR locus and ITP susceptibility (Table 2).17
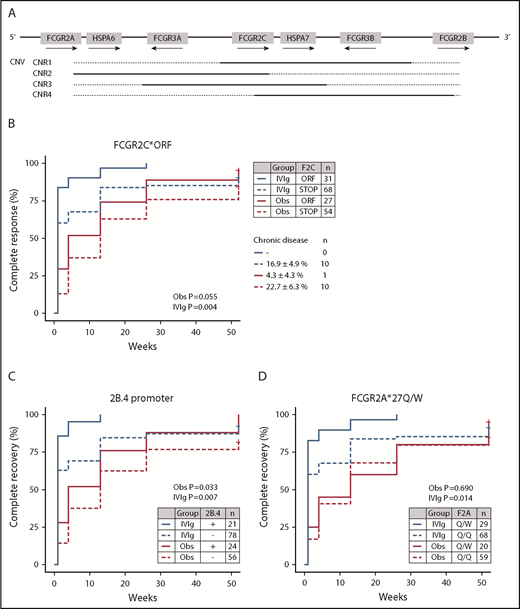
Association of FCGR2/3 variants with prognosis in newly diagnosed childhood ITP. (A) Overview of the human Fc γ receptor gene (FCGR) locus. CNV occurs in regions encompassing multiple genes (CNR). (B) Association of FCGR2C*ORF/STOP with complete recovery rates in patients that were carefully observed (Obs) or treated with IVIg. Complete recovery was defined by the International Working Group criteria as a platelet count of at least 100 × 109/L, which correlates strongly with absence of bleeding symptoms. FCGR2C*ORF enriched for children with favorable response to IVIg and a high rate of spontaneous recovery, and carried a low chance of chronic disease at 12 months’ follow-up. The FCGR2B/FCGR2C promoter variant 2B.4 (C) and FCGR2A*27Q/W SNPs (D), which are in linkage disequilibrium with FCGR2C*ORF, showed similar effects. In particular, FCGR2A*27Q/W did not differentiate as well between course of disease as the other 2 variants. P values are given for a log-rank test, stratified by treatment allocation.
Association of FCGR2/3 variants with prognosis in newly diagnosed childhood ITP. (A) Overview of the human Fc γ receptor gene (FCGR) locus. CNV occurs in regions encompassing multiple genes (CNR). (B) Association of FCGR2C*ORF/STOP with complete recovery rates in patients that were carefully observed (Obs) or treated with IVIg. Complete recovery was defined by the International Working Group criteria as a platelet count of at least 100 × 109/L, which correlates strongly with absence of bleeding symptoms. FCGR2C*ORF enriched for children with favorable response to IVIg and a high rate of spontaneous recovery, and carried a low chance of chronic disease at 12 months’ follow-up. The FCGR2B/FCGR2C promoter variant 2B.4 (C) and FCGR2A*27Q/W SNPs (D), which are in linkage disequilibrium with FCGR2C*ORF, showed similar effects. In particular, FCGR2A*27Q/W did not differentiate as well between course of disease as the other 2 variants. P values are given for a log-rank test, stratified by treatment allocation.
Sensitivity analyses of the TIKI cohort revealed an equal distribution of allele and haplotype variants in the full cohort compared with Caucasian patients only (available analyses for N = 135, showing 9% non-Caucasians; supplemental Table 3). Furthermore, odds ratios estimated on the full cohort and Caucasians only were similar.
Correlation of FCGR2/3 polymorphisms with recovery from newly diagnosed ITP
At present, no specific biomarkers are available to distinguish children who develop chronic ITP and children who have a transient course of disease; that is, with a spontaneous recovery or a favorable response to immunomodulatory therapy with IVIg. For association with prognosis we grouped patients from the TIKI trial who developed chronic ITP with the CINKID cross-sectional study. We observed that children with transient disease course (N = 131; N = 80 from the IVIg and 51 from the observation group, respectively) showed a significant enrichment of FCGR2A*27W, FCGR2C*ORF, and the 2B.4 promoter variant compared with patients with chronic ITP (Table 4), as well as healthy control individuals (supplemental Table 4). Only the association with FCGR2C*ORF remained significant when correction for multiple testing was performed. In particular, we observed that patients with FCGR2C*ORF showed a high likelihood of responding to IVIg and a higher rate of spontaneous recovery throughout 1 year follow-up (Figure 2B). None of 31 IVIg-treated patients and only 1 of 27 observed patients with FCGR2C*ORF showed chronic disease at 12 months’ follow-up (P = .043 compared with FCGR2C*STOP; Fisher’s exact test). The presence of an open reading frame allele in FCGR2C was associated with transient ITP with an odds ratio of 4.7 (95% CI, 1.9-14.3). Absolute recovery rates for patients with FCGR2C*ORF vs STOP are given in supplemental Table 5. The association of the FCGR2B/FCGR2C promoter variant 2B.4 and FCGR2A*27Q/W genotype with prognosis were similar to FCGR2C*ORF, but not as distinctive in particular for the FCGR2A*27Q/W genotype (Figure 2C-D). The odds ratio for transient ITP of the 2B.4 promoter was 2.6 (95% CI, 1.1-7.3), and that of FCGR2A*Q/W in comparison with FCGR2A*Q/Q was 3.3 (95% CI, 1.3-10.1). A similar skewing of variants was present for patients in the observation and IVIg group (supplemental Table 6).
Variants associated with transient ITP, compared with chronic ITP
| Transient ITP (self-limiting or IVIg-responsive) TIKI (N = 131) | Chronic ITP (CINKID & TIKI; N = 43) | P | P (FDR adjusted) | OR (95% CI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | ||||
| CNV | |||||||||||||
| CNR1 | 0 (0) | 4 (3) | 118 (90) | 9 (7) | 0 (0) | 0 (0) | 7 (16) | 33 (77) | 3 (7) | 0 (0) | .011 | .11 | 6.2 (1.8-24.7)* |
| CNR2 | 0 (0) | 0 (0) | 126 (96) | 5 (4) | 0 (0) | 0 (0) | 1 (2) | 42 (98) | 0 (0) | 0 (0) | .09 | .45 | |
| CNR3 | 0 (0) | 0 (0) | 131 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 43 (100) | 0 (0) | 0 (0) | ND | ND | |
| Alleles | |||||||||||||
| FCGR2A*27W | 88 (67) | 39 (30) | 4 (3) | 0 (0) | 0 (0) | 37 (86) | 5 (12) | 1 (2) | 0 (0) | 0 (0) | .041 | .37 | 3.3 (1.3-10.1)† |
| FCGR2A*131H | 27 (21) | 68 (52) | 36 (27) | 0 (0) | 0 (0) | 11 (26) | 18 (42) | 14 (33) | 0 (0) | 0 (0) | .50 | .37 | |
| FCGR2B*232T | 98 (75) | 30 (23) | 3 (2) | 0 (0) | 0 (0) | 31 (72) | 10 (23) | 2 (5) | 0 (0) | 0 (0) | .72 | 1.0 | |
| FCGR2C*ORF | 81 (62) | 48 (37) | 2 (2) | 0 (0) | 0 (0) | 38 (88) | 5 (12) | 0 (0) | 0 (0) | 0 (0) | .002 | .022 | 4.7 (1.9-14.3)‡ |
| FCGR2C*ncORF | 125 (95) | 3 (2) | 3 (2) | 0 (0) | 0 (0) | 38 (88) | 2 (5) | 3 (7) | 0 (0) | 0 (0) | .19 | .57 | |
| FCGR3A*158V | 46 (35) | 63 (48) | 22 (17) | 0 (0) | 0 (0) | 13 (30) | 22 (51) | 8 (19) | 0 (0) | 0 (0) | .87 | 1.0 | |
| FCGR3B*NA2 | 11 (8) | 66 (50) | 54 (42) | 0 (0) | 0 (0) | 9 (21) | 18 (42) | 16 (37) | 0 (0) | 0 (0) | .10 | .45 | |
| FCGR3B*SH | 128 (98) | 3 (2) | 0 (0) | 0 (0) | 0 (0) | 39 (91) | 4 (9) | 0 (0) | 0 (0) | 0 (0) | .06 | .37 | |
| FCGR2 promoter 2B.4 | 92 (70) | 39 (30) | 0 (0) | 0 (0) | 0 (0) | 37 (86) | 6 (14) | 0 (0) | 0 (0) | 0 (0) | .045 | .37 | 2.6 (1.1-7.3)‡ |
| Transient ITP (self-limiting or IVIg-responsive) TIKI (N = 131) | Chronic ITP (CINKID & TIKI; N = 43) | P | P (FDR adjusted) | OR (95% CI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | ||||
| CNV | |||||||||||||
| CNR1 | 0 (0) | 4 (3) | 118 (90) | 9 (7) | 0 (0) | 0 (0) | 7 (16) | 33 (77) | 3 (7) | 0 (0) | .011 | .11 | 6.2 (1.8-24.7)* |
| CNR2 | 0 (0) | 0 (0) | 126 (96) | 5 (4) | 0 (0) | 0 (0) | 1 (2) | 42 (98) | 0 (0) | 0 (0) | .09 | .45 | |
| CNR3 | 0 (0) | 0 (0) | 131 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 43 (100) | 0 (0) | 0 (0) | ND | ND | |
| Alleles | |||||||||||||
| FCGR2A*27W | 88 (67) | 39 (30) | 4 (3) | 0 (0) | 0 (0) | 37 (86) | 5 (12) | 1 (2) | 0 (0) | 0 (0) | .041 | .37 | 3.3 (1.3-10.1)† |
| FCGR2A*131H | 27 (21) | 68 (52) | 36 (27) | 0 (0) | 0 (0) | 11 (26) | 18 (42) | 14 (33) | 0 (0) | 0 (0) | .50 | .37 | |
| FCGR2B*232T | 98 (75) | 30 (23) | 3 (2) | 0 (0) | 0 (0) | 31 (72) | 10 (23) | 2 (5) | 0 (0) | 0 (0) | .72 | 1.0 | |
| FCGR2C*ORF | 81 (62) | 48 (37) | 2 (2) | 0 (0) | 0 (0) | 38 (88) | 5 (12) | 0 (0) | 0 (0) | 0 (0) | .002 | .022 | 4.7 (1.9-14.3)‡ |
| FCGR2C*ncORF | 125 (95) | 3 (2) | 3 (2) | 0 (0) | 0 (0) | 38 (88) | 2 (5) | 3 (7) | 0 (0) | 0 (0) | .19 | .57 | |
| FCGR3A*158V | 46 (35) | 63 (48) | 22 (17) | 0 (0) | 0 (0) | 13 (30) | 22 (51) | 8 (19) | 0 (0) | 0 (0) | .87 | 1.0 | |
| FCGR3B*NA2 | 11 (8) | 66 (50) | 54 (42) | 0 (0) | 0 (0) | 9 (21) | 18 (42) | 16 (37) | 0 (0) | 0 (0) | .10 | .45 | |
| FCGR3B*SH | 128 (98) | 3 (2) | 0 (0) | 0 (0) | 0 (0) | 39 (91) | 4 (9) | 0 (0) | 0 (0) | 0 (0) | .06 | .37 | |
| FCGR2 promoter 2B.4 | 92 (70) | 39 (30) | 0 (0) | 0 (0) | 0 (0) | 37 (86) | 6 (14) | 0 (0) | 0 (0) | 0 (0) | .045 | .37 | 2.6 (1.1-7.3)‡ |
Data are n (% of cohort). Bold values denote statistically significant P values. Chronic ITP was defined as absence of complete response (platelet count >100 × 109/L) 12 months after diagnosis. Transient ITP was defined as spontaneous recovery or a favorable response to IVIg 3 months after diagnosis. Patients from the TIKI trial with persistent ITP (recovery between 3 and 12 months) were excluded.
OR given for chronic ITP.
OR given for comparison of 27Q/W vs 27Q/W in association with transient ITP.
OR given for presence of 1 or 2 copies vs absence of the variant in association with transient ITP.
Finally, we investigated skewing of previously reported variants in chronic vs transient childhood ITP. The FCGR3A*158V allele was similarly distributed among the 2 patient groups and showed no association with clinical follow-up (Table 4). The FCGR2B*232I/I genotype, which is associated with early spontaneous recovery from childhood ITP,6 was not associated with prognosis beyond a 1-week follow-up (supplemental Figure 1 and Table 4).
Together, these results indicate that FCGR2C*ORF, FCGR2A*27W, and the 2B.4 promoter variant are associated with a transient ITP disease course and a favorable response to IVIg.
Correlation of a combined deletion of FCGR2C/FCGR3B with nonresponse to IVIg and development of chronic ITP
Patients with chronic ITP showed an increased frequency of a deletion of CNR1 compared with both children with transient ITP (Table 4) and healthy control individuals. Presence of the CNR1 deletion predisposed to chronic ITP with an odds ratio of 6.2 (95% CI, 1.8-24.7). Children with the CNR1 deletion had a significantly lower response rate to IVIg and a low spontaneous recovery rate in the observation cohort (Figure 3). Thus, these data suggest that presence of the CNR1 deletion is a prognostic indicator for prolonged disease courses and nonresponse to IVIg in childhood ITP.
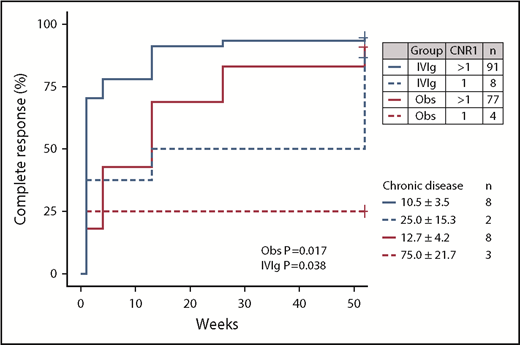
Effect of a deletion of CNR1 on complete recovery from newly diagnosed childhood ITP. Complete recovery was defined by the International Working Group criteria as a platelet count of at least 100 × 109/L. P values are given for a log-rank test stratified per treatment group.
Effect of a deletion of CNR1 on complete recovery from newly diagnosed childhood ITP. Complete recovery was defined by the International Working Group criteria as a platelet count of at least 100 × 109/L. P values are given for a log-rank test stratified per treatment group.
Discussion
The FCGR2/3 locus is highly influential in regulating traits of the human immune system, and multiple genetic polymorphisms of the locus are significantly associated with the development and severity of autoimmune diseases.11,35 Using a unique cohort of children with newly diagnosed ITP who were followed longitudinally for 1 year, as well as a second cohort of children with chronic ITP, we determined the prognostic significance of FCGR2/3 polymorphisms. We found a strong association of FCGR2C*ORF with susceptibility to ITP, which is in linkage disequilibrium with FCGR2A*27W and the 2B.4 promoter variant. The same polymorphic markers also correlated with transient disease courses, as opposed to chronic disease, and a favorable response to IVIg. Children who progressed to chronic ITP showed an increased frequency of the CNR1 deletion, which was also associated with a failure to respond to IVIg treatment. We conclude that patients with transient childhood ITP and those who develop chronic ITP show skewed FCGR2/3 polymorphisms relative to each other.
Children with newly diagnosed ITP and mild bleeding symptoms are routinely managed by observation alone.36 Nevertheless, the disease exerts a significant effect on children and their families.37,38 Our data suggest that genotyping of FCGR2C*ORF could indicate a transient ITP course, and these children also have a high probability of responding to IVIg. Furthermore, determination of CNV in CNR1 could help to identify patients with a predisposition for prolonged thrombocytopenia. Together, this may be helpful in determining prognosis of newly diagnosed childhood ITP for clinical counseling, and may indicate the likelihood of response to IVIg if treatment is intended.
The identified FcγR variants may have an effect on multiple levels of the immune response, including antigen presentation and regulation of the immune response, as well as clearance of antibody-opsonized platelets. Given the linkage disequilibrium among FCGR2A*27W, FCGR2C*ORF, FCGR3A*158V, and 2B.4, it remains unresolved which genetic variants are causative for the observed associations. Our study had insufficient power to test the linkage of haplotypes with susceptibility to ITP. However, based on functional consequences of these variants, we can infer potential mechanisms. FCGR2A*27W has no effect on receptor expression among various immune cells and does not alter antibody-dependent cellular cytotoxicity, making it less likely that this variant is causative.39 Although we observed no difference in the frequency of rare FCGR2A*27W/W homozygotes in ITP, the aforementioned functional data do not support a nonlinear allele effect. In contrast, FCGR2C has long been considered a pseudogene because of a premature stop codon.40,41 We now know that in individuals with the variant FCGR2C*ORF, FcγRIIc is expressed by neutrophils, monocytes, and NK cells.25,42 Macrophages and B cells may also express FcγRIIc.11,13,43 Expression of FcγRIIc may result in enhanced phagocytic and antibody-dependent cellular cytotoxicity activity or impaired downregulation of B-cell responses, and thereby a predisposition to ITP. In particular, individuals with FCGR2C*ORF show evidence of enhanced humoral immune responses.13,44 In contrast, presence of the FCGR2B/2C promoter variant 2B.4 alters the expression levels of FcyRIIb40,45 and has been associated with systemic lupus erythematosus,27,40 as well as response to IVIg treatment in patients with Kawasaki disease.46,47 In sum, these FCGR2/3 polymorphisms may facilitate an increased innate immune phagocytosis or antibody-dependent cellular cytotoxicity, or enhanced adaptive immune responses that result in a dysregulated immune response and transient ITP. The transient character of thrombocytopenia could be a result of a delayed regulation of this enhanced immune response. Here, IVIg treatment would contribute to the restoration of this dysregulated immune response.
The identification of an overrepresentation of a CNR1 deletion in patients with chronic ITP and an association with treatment response was striking. The deletion of CNR1 concerns an 82-kb region encompassing both FCGR2C and FCGR3B genes, and may extend into the promoter region of FCGR2B (Figure 2A), leading to an altered expression pattern of FcγRIIb by NK cells.25 A deletion of FCGR3B has been extensively associated with systemic lupus erythematosus susceptibility and disease severity.27,28,48,49 The reduced expression of FcγRIIIb resulting from low copy numbers of the gene has been associated with impaired clearance of immune complexes,48 which may explain the enhanced systemic lupus erythematosus phenotype. Furthermore, the deletion changes the expression pattern of FcyRIIb, as previously shown,25 which may serve as an alternative explanation for the observed effect by altering the threshold to mount immune responses. Although FCGR3A*158V has been associated with susceptibility to childhood ITP,17 this variant did not correlate with development of chronic ITP, which is in accordance with earlier reports.16,19,20 Taken together, an impaired cellular response to immune complexes could be an initial induction event that results in chronic ITP, possibly mediated by polymorphonuclear or NK cells. In such a situation, the combination of prolonged inflammation, tissue damage, and priming and activation of antigen-presenting cells could also enhance epitope spreading, leading to platelet autoantibody formation.50
The generalizability to newly diagnosed ITP is largely defined within the context of the TIKI trial’s inclusion criteria; most important, a presenting platelet count of 20 × 109/L or less, and absence of severe or life-threatening bleeding that required medical treatment. Patients included from the CINKID study had similar baseline characteristics, including age and rate of preceding infections. Furthermore, the results cannot be extended beyond a Caucasian population. We suggest that independent validation of this study’s data should be performed before genotyping could be used to determine prognosis.
This is the largest genetic association study in childhood ITP to date, but despite this, the available sample size imposed limits on our ability to perform haplotype linkage analyses, and also showed low power during multiple comparison correction. For susceptibility to childhood ITP, we confirmed our findings by meta-analyses. For CNR1, we suggest that extensive epidemiological and biological data associate this variant with susceptibility and severity of other autoimmune diseases,27,28,48,49 yet we are the first to analyze this variant in the context of chronic childhood ITP, and our findings remain to be confirmed in an independent cohort.
In conclusion, we identified genetic risk factors in the FCGR2/3 locus that are clearly associated with susceptibility to self-limiting ITP and a favorable response to IVIg. In addition, the increased susceptibility to chronic ITP conferred by the CNR1 deletion shows that genetic autoimmune susceptibility variants may play a more important role in ITP than previously acknowledged. Collectively, the data highlight the complexity of the possible phenotypes resulting from genetic variation within the FCGR2/3 locus. Our findings indicate that targeted genotyping of the FCGR2/3 locus may be useful in determining prognosis of childhood ITP, and could potentially inform treatment decisions.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors are grateful to Saidyliëne Concepcion and Tamara Stegmann for technical assistance. The authors thank Michael W. Tanck from the Academic Medical Center, Amsterdam, and Marjanka Schmidt from the Netherlands Cancer Institute, Amsterdam, The Netherlands, for advice regarding statistical analyses.
This work was supported by a research grant from the Landsteiner Foundation for Blood Transfusion Research and a doctoral stipend to D.E.S. by the Studienstiftung des Deutschen Volkes.
Authorship
Contribution: D.E.S. analyzed and interpreted data and wrote the manuscript; K.M.J.H.-P. analyzed and interpreted data and designed the clinical studies; A.G.L. contributed to clinical studies and analyzed and interpreted data; M.C.A.B. designed the clinical studies and contributed to the design of the study; B.V. and S.Q.N. performed experiments and analyzed and interpreted data; T.W.K., L.P., and C.E.v.d.S. discussed data; G.V. designed and supervised the study and wrote the manuscript; M.d.H. designed and supervised the study, designed the clinical studies, and wrote the manuscript; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Masja de Haas, Department of Immunohematology Diagnostics, Sanquin Diagnostic Services, PO Box 9190, 1006 AD Amsterdam, The Netherlands; e-mail: m.dehaas@sanquin.nl.

