

Abstract
Unveiling the mechanisms and the cellular dynamics at the basis of human hematopoietic homeostasis has been a main focus for the scientific community since the discovery of a pool of multipotent hematopoietic stem cells (HSCs) capable of sustaining the hematopoietic output throughout life and after transplantation. Recently, new works shed light on the (1) differentiation paths, (2) size and replication rate of human HSC population at steady state, and (3) role of the distinct subpopulations comprising the hematopoietic stem and progenitor cell reservoir after transplantation. These papers exploited cutting-edge technologies, including vector integration site clonal tracking, spontaneous mutations, and deep transcriptome profiling. Here we discuss the latest updates in human hematopoietic system biology and in vivo dynamics, highlighting novel concepts and common findings deriving from different approaches and the future directions of these studies. Taken together, this information contributed to partially resolving the complexity of the in vivo HSC behavior and has major implications for HSC transplantation and gene therapy as well as for the development of future therapies.
Introduction
The hematopoietic system represents a unique dynamic organization in which short-term living cells, with diverse properties and functions, require continuous production from a pool of hematopoietic stem/progenitor cells (HSPCs), mainly resident in the bone marrow (BM) during adult life. From the very beginning, the discovery of hematopoietic stem cells (HSCs) has lead to their exploitation in the clinical setting for transplantation and, more recently, gene therapy (GT) with the aim of treating innate or acquired disorders.1-4 Despite their wide and established clinical use, several aspects related to their functional properties in humans have yet to be clarified.
The identification of the human HSPC population was classically based on the use of the CD34 cell surface marker, which is used to enrich HSPCs in the context of transplantation and GT. During the last few years, the complexity of the HSPC compartment has been revealed, and novel subsets have been described thanks to the use of specific surface markers (CD38, CD90, CD45RA, CD49f, CD71, CD41, CD10, CD7, CD135) that allow the identification of both primitive multipotent HSPC subsets and “common/committed” progenitors with specialized differentiating potential and distinct long-term (LT) survival.5-7 The functional properties and hierarchical relationships with other HSPC subpopulations of each novel subset were mainly tested exploiting single-cell in vitro assays6-9 and xenotransplantation animal models.10-12 Additional differentiation systems exploiting induced-pluripotent stem cells13,14 and 3-dimensional scaffolds15 have been proposed to study the clonal output of human HSPCs in vitro.
Although representing efficient strategies for testing the differentiation potential of newly identified subpopulations, these experimental settings are not exempt from intrinsic limitations, still leaving open the debate on the actual in vivo frequency, functionality, and longevity of HSPC clones in humans. Moreover, there is still a large debate on the actual landscape of human hematopoiesis in vivo also due to the methodological difficulties in studying real-time human hematopoiesis.
The application of novel single-cell methods and the possibility of tracking permanent/acquired genomic modifications to better investigate the human hematopoietic system permitted a reshaping of the boundaries among HSPC subpopulations, measurement of HSC population size and differentiation rate, and assessment of the fate of HSPC subpopulations after transplantation.
HSPC differentiation and hierarchy
Historically, blood formation was thought to occur through stepwise progression from HSC to oligo-, bi-, and unipotent progenitors following a strict hierarchical structure (Figure 1). In this “classical model,” differentiation of self-renewing and multipotent HSCs is associated with loss of “stem” properties and acquisition of lineage-specific potential that generates single lineage-restricted progenitors. According to this hierarchical structure, HSCs directly differentiate into multipotent progenitors (MPPs) before moving to lymphoid/myeloid commitment determination. This model was hypothesized after the identification of phenotypically distinct progenitors with lineage-restricted potential in the mouse setting,8,16 and it represented the major frame of reference also for its human counterpart. However, many publications challenged this classical view showing that lineage choice occurred in the primitive HSC compartment10,17,18 and that lymphoid/myeloid fates remained coupled along differentiation after loss of erythroid potential.19
Although much effort has been made to clarify murine hematopoiesis, the applicability of this information in the human setting remains to be assessed. Moreover, the transcriptional networks regulating HSC emergence, self-renewal, and differentiation were mainly analyzed starting from predefined populations, sorted according to the expression of specific surface markers, without considering the transcriptional heterogeneity in each subset and the transitional window between 2 cell states.
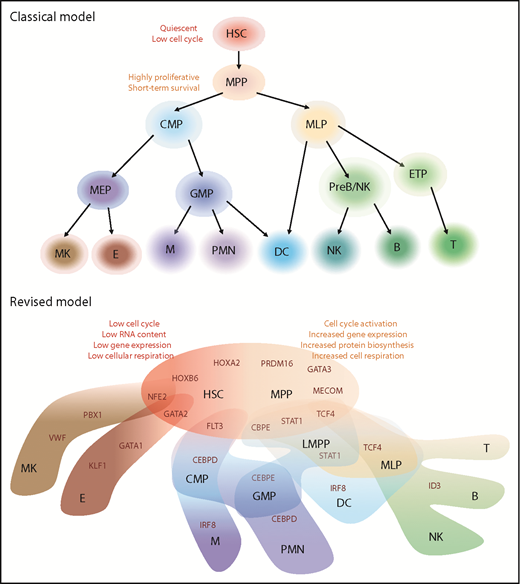
Models of human hematopoietic hierarchy. Representation of classical and revised models for HSC differentiation toward mature lineages based on recent literature.22-25 We reported in black the name of hematopoietic populations, whereas in dark red, the differentiation-driving genes. HSCs and MPPs shared similar HOXB6/HOXA2/PRDM16 gene modules but different metabolic state. HSC characteristics are listed on top left (in red), whereas MPP state is associated with activation of mechanisms listed on top right (orange). In the HSC/MPP cloud, preexisting lineage-specific modules are present at low levels and reinforced along differentiation: NEF2/GATA2 module represents the first attraction point for the megakaryocytic/erythroid specification, whereas CBPE/STAT1/TCF4 is associated with lymphoid-myeloid primed progenitor (LMPP) specification. Interestingly, dendritic cell (DC) and multilymphoid progenitor (MLP) cell fates appear to be associated with STAT1 expression, but IRF8 and ID3/TCF4 modules drive their final specification, respectively. The link between MLP and DC is in line with previous publications.9 B, B cells, CMP, common myeloid progenitors; E, erythrocytes; ETP, early T-cell progenitors; GMP, granulocytes monocytes progenitors; M, Monocytes; MEP, megakaryocytes erythrocytes progenitors; MK, megakaryocytes; NK, natural killer cells; PMN, polymorphonucleated cells; PreB/NK, B-cell and NK cell progenitors; T, T cells.
Models of human hematopoietic hierarchy. Representation of classical and revised models for HSC differentiation toward mature lineages based on recent literature.22-25 We reported in black the name of hematopoietic populations, whereas in dark red, the differentiation-driving genes. HSCs and MPPs shared similar HOXB6/HOXA2/PRDM16 gene modules but different metabolic state. HSC characteristics are listed on top left (in red), whereas MPP state is associated with activation of mechanisms listed on top right (orange). In the HSC/MPP cloud, preexisting lineage-specific modules are present at low levels and reinforced along differentiation: NEF2/GATA2 module represents the first attraction point for the megakaryocytic/erythroid specification, whereas CBPE/STAT1/TCF4 is associated with lymphoid-myeloid primed progenitor (LMPP) specification. Interestingly, dendritic cell (DC) and multilymphoid progenitor (MLP) cell fates appear to be associated with STAT1 expression, but IRF8 and ID3/TCF4 modules drive their final specification, respectively. The link between MLP and DC is in line with previous publications.9 B, B cells, CMP, common myeloid progenitors; E, erythrocytes; ETP, early T-cell progenitors; GMP, granulocytes monocytes progenitors; M, Monocytes; MEP, megakaryocytes erythrocytes progenitors; MK, megakaryocytes; NK, natural killer cells; PMN, polymorphonucleated cells; PreB/NK, B-cell and NK cell progenitors; T, T cells.
Recent developments in single-cell technologies have now enabled high-throughput genome, epigenome, and transcriptome analyses of single cells20 for studying cellular compartment heterogeneity. In particular, several groups have applied single-cell RNA sequencing (scRNAseq) to human hematology, providing unprecedented insights into the complexity of phenotypically defined compartments, in cellular differentiation hierarchy as well as the overall gene regulatory networks.21
One of the novel concepts challenging the classical view of hematopoietic differentiation is that primitive HSPCs appear to gradually acquire lineage biases along multiple directions, rather than passing through discrete hierarchically organized progenitor populations.22,23 In particular, novel data support the idea that primitive HSC and MPP subsets display an “unspecialized” cell status with different degrees of cell-cycle activity and low levels of early lineage-priming gene sets that compete for ultimately directing them toward a specialized cell fate (Figure 1).
By integrating flow cytometric, transcriptomic, and functional data at the single-cell level, Velten et al22 analyzed the BM HSPC compartment to map early differentiation of human HSCs. According to the proposed model, unilineage-restricted cells emerge directly from a “continuum of low-primed undifferentiated hematopoietic stem and progenitor cells,” defined as “CLOUD-HSPC.” In the CLOUD, lympho/myeloid and megakaryocytic/erythroid specifications represented the major routes of lineage priming, even when a clear separation into single lineages was not present. In this novel view, differentiation should be considered a transitory state within HSPC continuum with a higher probability of commitment to particular lineages. This “flow of differentiation” was also hypothesized using a completely different approach. The combination of single-cell epigenomic profiling through Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) with scRNAseq on 10 phenotypically defined HSPC subsets allowed the association of changes in transcription factor expression to changes in chromatin accessibility toward differentiation.23 These results suggested that the chromatin accessibility landscapes in early hematopoietic differentiation comprise a broad pool of allowable states, similar to the CLOUD-HSPC described by Velten et al,22 and continuous differentiation trajectories direct to lineage specifications.
Interestingly, similar gene modules were associated with the direction/degree of priming in the 2 data sets. In particular, the least-primed state was characterized by expression of HOX motif, cell-cycle quiescence, and low RNA content, whereas the master lineage regulators ID3, CEBP, and GATA1 show gradients of activity across lymphoid, myeloid, and erythroid development, respectively (Figure 1). During continuous priming and differentiation process, stem cell modules and some early priming motifs already expressed in primitive HSC are turned off concomitantly, reinforcing lineage-specific modules that drive the final lineage commitment.
Two additional studies dissected the cellular hierarchy and heterogeneity of lymphoid/myeloid-24 and megakaryocyte/erythrocyte-committed25 HSPC progenitors. A new gating strategy based on CD45RA and CD10 markers for the isolation of human cord blood progenitors with myeloid-only (GMP), lymphoid-only (MLP), and combined myeloid/lymphoid (lymphoid-myeloid primed progenitors) potential has been proposed, and the use of CD44, CD41, and CD71 markers allows the definition of erythrocyte/megakaryocyte bipotent (Pre-MEP) and biased (E-MEP and MK-MEP) progenitors in the mobilized peripheral blood (MPB)–derived megakaryocyte/erythrocyte progenitor (MEP) compartment. Despite focusing on defined lineage-primed progenitor compartments, these 2 studies also implied that differentiation is a continuum rather than a subsequent binary branching decision process.
However, current scRNAseq technologies still have some limitations. The ability to detect only highly expressed genes, the discrepancy between messenger RNA levels and protein expression, and the presence of many shared biological processes (such as cell cycle, metabolism) could generate substantial heterogeneity. Finally, although providing novel insights on the molecular pathways activated during human hematopoietic lineage differentiation, scRNAseq data still rely on single-cell culture and xenotransplantation for functional validation. As already mentioned, these experimental approaches are not able to fully recapitulate physiological in vivo differentiation, highlighting the need to exploit alternative strategies to study the HSPC dynamics directly in humans.
In vivo dynamics of human HSC in unperturbed system
Assessing the HSPC dynamics in an unperturbed BM environment directly in humans represents a methodological challenge. In vivo color labeling and transposase-based inducible systems have been used to estimate the replication rate of murine HSCs26-29 in vivo. These experimental settings are mostly inapplicable to humans. Thus, in past years, novel approaches based on unique naturally occurring genomic modifications, such as the shift of 50:50 X-chromosome inactivation ratio,30 telomere length distribution,31 and spontaneous mutations,32,33 have been progressively employed to address in vivo behavior of human HSCs.
As a result of X-chromosome inactivation during embryogenesis, somatic cells in women maintain only 1 of the 2 X chromosomes active. Because this process occurs randomly, the ratio of HSCs with maternal vs paternal X chromosome should be ∼50:50. However, with aging, this ratio progressively changes due to different expansion, differentiation, and exhaustion of clones. Thanks to this natural tendency, X-inactivation has been used as a marker for studying the dynamics of human HSPCs. In the same way, the occurrence of spontaneous mutations in the stem cell pool and the shortening of telomeres due to cell divisions enable the quantification of the number, the activity, and the longevity of human HSCs. Although leading to some common conclusions, these strategies display some discrepancies in quantifying the number of stem cells actively contributing to the hematopoiesis and the rate of their cell divisions.
There is general agreement on the dynamics of the stem cell pool during hematopoietic maturation and aging (Figure 2). It is now well established that the hematopoietic maturation is accompanied by expansion of the HSC pool from birth to adolescence, and relatively stable HSC numbers characterize adulthood. This 2-phase hematopoiesis may be the result of increased physical space in the BM niches due to bone size growth during adolescence, combined with the concept that HSC replication in mammals’ lifetime is finite and conserved. A limited replication capacity and activation of senescence mechanisms would help to preserve genetic integrity.
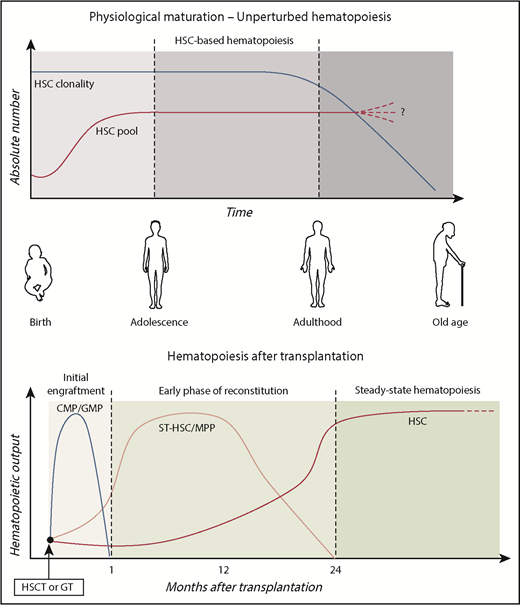
Human HSPC dynamics in vivo. Schematic representation of the role of primitive and committed HSPC subpopulations during physiological aging and after transplantation. HSC pool increases in size during childhood, reaching the final adult reservoir that allows maintenance of hematopoietic production during life. As results of continuous challenging from external stimuli (such as infections, environmental pollution, and radiations), the HSC pool progressively loses its clonal complexity. Although previous works described increased frequency of HSC pool in the elderly,58,59 a comprehensive assessment of the maintenance, increase, or decrease of HSC number during aging remains to be elucidated. After transplantation, hematopoietic output is sustained by different HSPC subsets over time.49 In the initial engraftment, myeloid production is sustained by short-living myeloid progenitors (CMP/GMP). Short-term HSC/MPPs activated in the early phases provided the first hematopoietic reconstitution. Around 1 to 2 years after transplant, the HSPC compartment stabilized and LT HSCs maintained steady-state hematopoiesis.
Human HSPC dynamics in vivo. Schematic representation of the role of primitive and committed HSPC subpopulations during physiological aging and after transplantation. HSC pool increases in size during childhood, reaching the final adult reservoir that allows maintenance of hematopoietic production during life. As results of continuous challenging from external stimuli (such as infections, environmental pollution, and radiations), the HSC pool progressively loses its clonal complexity. Although previous works described increased frequency of HSC pool in the elderly,58,59 a comprehensive assessment of the maintenance, increase, or decrease of HSC number during aging remains to be elucidated. After transplantation, hematopoietic output is sustained by different HSPC subsets over time.49 In the initial engraftment, myeloid production is sustained by short-living myeloid progenitors (CMP/GMP). Short-term HSC/MPPs activated in the early phases provided the first hematopoietic reconstitution. Around 1 to 2 years after transplant, the HSPC compartment stabilized and LT HSCs maintained steady-state hematopoiesis.
Data derived from the analyses of X-chromosome ratio drift with aging in blood cells of 1226 female humans30 and of the telomere shortening in peripheral blood granulocytes in 356 healthy subjects31 allowed estimation of a similar rate of human HSC replication of ∼1 per year during adult life. Based on this estimate, it was calculated that ∼1275 HSC-derived clones contributed to human hematopoiesis and maintain steady-state blood cell production. This number is intriguingly similar to data derived from clonal tracking studies in GT-treated patients, exploiting vector integration sites (ISs) as markers for monitoring the dynamics of hematopoietic reconstitution after infusion of gene-corrected cells.34
These findings have been recently challenged by 2 papers exploiting spontaneous mutations to study in vivo dynamics of HSCs.32,33 Although the first 2 studies30,31 relied on data from bulk populations, Osorio et al’s33 and Lee-Six et al’s32 works were based on in vitro expansion of single sorted HSPCs from healthy donors followed by whole-genome sequencing. Through this strategy, it was estimated that the rate of mutations is ∼14 base substitutions per year, and the vast majority were acquired after birth. Very few mutations were found to be shared among the primitive HSPCs analyzed, supporting the idea that the genomic modifications detected represent independent events occurring at the single-cell level. Nevertheless, rare common mutations were also found in nonhematopoietic cells, allowing the creation of a phylogenetic tree starting from events occurring in the embryo. Approximate Bayesian computation framework applied on simulated and experimental data allowed the estimation of the time between successive self-renewal stem cell divisions, which was in the range of 2 to 20 months, and the number of HSCs actively contributing to circulating granulocytes, and thus to hematopoiesis, which was in the range of 44 000 to 215 000. These conclusions are clearly in contrast with the definition of “HSC-status” characterized by quiescence, low cell-cycle activity, and slow differentiation rate. As pointed out by the authors, the limited amount of total cells analyzed (140 in the work of Lee-Six et al32 and 18 in the work of Osorio et al33 ) of healthy donors (only one 59-year-old man32 and 5 donors,33 respectively) and the possible overestimation of mutation rate introduced by ex vivo culture could explain the differences in the results obtained compared with the past literature. A recent work showed how these limitations can be overcome by analyzing somatic mutations in mitochondrial DNA (mtDNA) retrieved from existing scRNAseq or assay for transposase-accessible chromatin using ATAC-seq.35 This novel approach was applied to assess hierarchical clonal relationships in several tissues and cell types showing high sensitivity and large applicability. Moreover, it has the main advantage of being immediately available, because the mtDNA mutations can be detected by commonly employed single-cell sequencing. This promising approach has enlarged the toolkit available to researchers for human in vivo studies, combining clonal tracking data with epigenomic and transcriptomic state of single cells.
However, genomic mutations have provided important information on the functional properties of stem cells in humans. Although previous studies of unperturbed hematopoiesis in mice suggested that hematopoiesis was predominantly sustained by MPPs,26,27 several publications exploiting numerous in vivo labeling systems have now proven that murine adult HSCs robustly contributed to steady-state hematopoiesis.18,28,29 The exploitation of somatic mutations in humans has led to the proposal that phenotypically defined HSCs actively contribute to steady-state hematopoiesis also in humans (Figure 2). This conclusion, together with the data on transplanted human HSPC discussed below, adds a new frame of reference for future data interpretation.
Role of human HSPC after transplantation
Despite the progress toward the characterization of true repopulating stem cells by specific features, such as cell surface phenotype and cycling characteristics, the only assay able to define and unequivocally study these cells is the engraftment and repopulation of all hematopoietic lineages after transplantation. As mentioned above, the capability of replenishing all mature blood cell lineages maintaining self-renewal competence and LT survival led to the clinical exploitation of HSCs. Nowadays, hematopoietic stem cell transplantation (HSCT) remains the main curative treatment of many hematological malignancies and for inherited genetic disorders,1,2 together with HSPC-based GT.3,4,36-41 In this context, understanding the role of HSPC subpopulations after transplantation is of paramount importance for the improvement of clinical procedures aimed at establishing not only LT maintenance of the graft but also early recovery from cell aplasia.
In the past years, novel approaches based on in vivo cell tracking exploiting stable and unique clonal markers have been progressively employed to address the behavior of HSPCs after transplantation.42 In this context, the analysis of viral ISs has emerged as one of the most prominent strategies allowing the tracking of the activity of genetically engineered hematopoietic cells not only in animal models11,18,26,27,43-45 but also in GT-treated patients.34,38,46-49 Upon stem cell transduction, each cell and its progeny become univocally marked by a specific IS. Retrieving ISs from mature blood cells after stem cell transplantation has allowed the study of the kinetics of blood cell production from individual stem cells within a heterogeneous population. IS analyses have the potential to allow the assessment of (1) the number of active HSC and the clonal output of HSPC during different phases of hematopoiesis (early reconstitution and steady state); (2) the survival of committed progenitors; and (3) the modeling of the hierarchy of human hematopoiesis by analyzing the shared ISs among different lineages.
In the murine setting, the finding that the vast majority of the ISs after transplant were present in either lymphoid or myeloid cells with few ISs shared by both lineages,19,43 together with data derived from transplanted single HSC clones,17,50 led to the concept that murine HSCs are heterogeneous and already biased toward their fate. Recently, with the exploitation of a mouse model bearing fluorescent protein cassettes under the control of Gata1 and von Willebrand Factor (Vwf) lineage-specific promoters, megakaryocytes/erythrocytes–restricted HSCs with LT survival were described.51 Interestingly, despite transplanting >1000 mice with single LT HSCs, the authors did not find evidence of other types of lineage-restricted HSCs at any time point, a different result from previous data. However, mouse strain specificity of competitor cells, very low levels of engraftment, and type of conditioning (irradiation vs drug administration) could be responsible for these discordant results. To take into consideration all these variables, the Weissman group performed a comprehensive barcoding study assessing lineage commitment at the single-clonal level in (1) wild-type vs immune-deficient mice; (2) unconditioned vs conditioning treatments; (3) high vs low dose of helper cells.52 This impressive data set suggests that hematopoietic differentiation occurs differently under steady-state and stressed conditions with lineage-biased LT-HSC clones arising only in perturbed systems.52
Clonal tracking studies in nonhuman primates have been pivotal in studying HSCT dynamics in an experimental setting close to humans.11,44,45 The results of these works showed common patterns of hematopoietic reconstitution upon transplantation: clonal fluctuation in the early phases postinfusion of marked HSPC, potentially due to the initial contribution to the hematopoiesis of short-term unilineage progenitors, followed by a recovery of a stable hematopoietic output likely related to the takeover of LT multipotent HSC contribution. LT HSCs are able to provide multilineage engraftment, and there is currently no evidence of predetermined lineage choice at the stem cell level in primates.
The increasing amount of data generated during these studies and the need to combine multiple types of information (cell phenotype and absolute count, number of ISs and their clonal abundance, percentage of transduction, and number of vector integrated copies in each subpopulation) reinforced the use of mathematical models for measuring and statistically validating descriptive kinetics of hematopoietic reconstitution.53 The application of mathematical models to data sets deriving from clonal tracking studies in nonhuman primates not only proved that random sampling alone cannot explain the observed initial clone fluctuations but also suggested that the main driver of this behavior resides in the limited number of progenitor cell divisions before exhaustion. Moreover, analyzing granulocytes dynamics as markers for HSC output after establishment of steady state, the total HSC differentiation rate was calculated to be around 1 every 200 days, very similar to previous estimations.53 Thus, IS-based tracking studies coupled with mathematical models are able to provide novel information on the hematopoietic compartments at steady state.
Subjects treated with genetically repaired HSPCs represent a unique source of data to study human hematopoiesis and dissect kinetics of hematopoietic reconstitution upon transplantation, because a substantial fraction of the engrafting HSPC and their differentiated progeny are marked by the therapeutic vector in a unique genomic site.42 To date, few cutting-edge studies have exploited ISs retrieval from GT-treated patients, allowing the study of the complexity of hematopoietic system and hematopoietic reconstitution upon HSCT in humans.34,47,49,54 A key prerequisite is that hematopoiesis is reconstituted from multiple clones, and there is no skewing for insertion sites near potential genotoxic sites.42,55 Longitudinal analyses on HSC-GT patients unveiled that unilineage clones active during the first 6 months after GT tend to be replaced by multilineage LT clones, indicating HSC-derived activity.34,47 Within the BM CD34+ cell population, the number of clones was higher in the early post-GT phase and decreased over the following months to a smaller steady-state population of ∼1200 clones contributing actively to the hematopoiesis,34 in line with previous findings.30 Recently, our group further dissected HSPC compartment dynamics by isolating 7 distinct HSPC subsets at different time points after transplantation.49 Our data show that stabilization of HSPC compartment composition occurs ∼1 to 2 years after transplantation, suggesting that the establishment of the steady-state hematopoiesis required more time with respect to the originally proposed 6 months. Probabilistic models and sharing of ISs with mature lymphoid and myeloid populations uncovered, for the first time, that human primitive HSPCs have a distinct role in sustaining hematopoiesis after transplantation. Although MPPs are more active in the early phases, long-living HSCs are at the top of the hematopoietic hierarchy at steady state (Figure 2). Finally, although myeloid committed progenitors required continuous production from upstream MPP/HSC populations, the lymphoid-specific IS marking of MLPs implied a higher HSC-independent survival capability for lymphoid progenitors. This latter finding can be also explained by the existence of a pool of lymphoid-biased LT-HSC, specifically supplying MLP production, as previously suggested by other groups.56 However, it should be considered that the vast majority of these works were based on data derived from GT trials for primary immunodeficiencies in which the effect of selective advantage for corrected cells could have influenced the dynamics of reconstitution in specific cell compartments. In addition, the disparities in conditioning regimens57 and of stem cell sources in the distinct clinical trials may account for differences. Specific and comprehensive study design would be required in order to consider all the possible variables and draw definitive conclusions.
ISs were also cleverly exploited to map regulatory regions (enhancers and promoters) active in LT HSC during transduction. γ-Retroviral (γ-RV) vectors stably integrate into the cell genome with a substantial preference for strong enhancer and active promoters, thus permanently marking active regions at the moment of transduction.48 By collecting ISs from 10 LT γ-RV HSPC-based GT-treated patients, it was possible to map >3000 gene regulatory regions active in LT HSCs. Of note, novel microRNAs (miR-10a and miR-335) regulating HSC fate and differentiation, in particular lymphopoiesis, were identified. Although performed on a cohort of patients with a high rate of leukemia due to insertional mutagenesis and the caveat that chromatin accessibility landscape could have been influenced by in vitro cytokine stimulation required for γ-RV transduction, this work showed an innovative approach for the detection of regulatory regions in functionally defined LT HSCs in humans.
These reports represent prototypical examples of the power of translational studies, providing information relevant to the human hematopoietic system, complementing and expanding the data derived from animal models.
Aging of human HSCs
Because of extended global life expectancy, many research groups have focused their attention on assessing the mechanisms and processes involved in the aging of human hematopoiesis.58,59 Accumulation of DNA damage during cell proliferation,60,61 telomere shortening, and inflammation62 act on the HSC compartment, triggering a plethora of cellular programs leading to senescence.63 These cellular pathways are also activated during BM transplantation. Indeed, upon infusion of donor cells, HSPCs undergo strong proliferation because of the need to repopulate the host BM and reconstitute the hematopoietic system. This proliferative stress would explain the accelerated aging of hematopoietic compartment in LT BM-transplanted patients.64 Thus, understanding the mechanisms acting on senescent HSC could allow the discovery of novel compounds to increase the fitness of stem cells with a direct implication on transplantation LT outcome.65
From a clinical point of view, human hematopoietic aging is accompanied by reduced immune functions, anemia, and increased tendency of developing malignancies. Age-associated changes in HSPC compartment have been extensively characterized in mice. Recently, reduction of HSC clones and lymphoid potential was observed in HSPC compartments of aged vs young mice through in vivo labeling of HSPC.66 Interestingly, stress induced by serial transplantations resulted in clonal collapse in both groups of animals, implying that continuous challenging of the hematopoietic compartment during the entire life of a subject would be one of the major drivers of HSPC aging.
However, given the different life span of rodents with respect to primates, the use of these notions in the human context has yet to be defined. By analyzing >3000 somatic mutations of unusual allelic fractions on peripheral blood samples collected from 12 380 individuals, it was assessed that 10% of subjects older than 65 years old showed clonal hematopoiesis.67 This phenomenon is increasingly common as people age and is associated with a higher risk of developing hematological malignancies. Main drivers of clonal expansion were identified in DNMT3A, ASXL1, and TET2 mutations. These mutations, occurring in apparently healthy subjects, may represent characteristic early events in the development of cancer.
Progressive decline of clonal diversity and pronounced expansion of both myeloid and B-cell lineage–restricted clones were also described in 2 aged rhesus macaques transplanted with autologous transduced HSPC.68 Both aged animals showed a gradual decrease in the number of clones contributing to hematopoiesis, suggesting an increased exhaustion rate in comparison with young transplanted animals. However, in addition to the low number of animals involved in the study, it should be underlined that possible preexisting alterations of the HSPC compartment and the effect of replication stress induced by the transplantation could have exacerbated the hematopoietic phenotype observed in the aged animals. Nevertheless, this strategy proved useful to study the effect of clonal hematopoiesis in aged primates and represents a step forward in the study of hematopoietic aging in a context closer to humans.
Conclusions and final remarks
The considerable technological advancements made during the past years have led to increasing our understanding of hematopoietic dynamics in humans. Despite the growing amount of information generated from different innovative single-cell approaches, technical limitations are still present. scRNAseq data have now pointed out the need to redefine our phenotypic characterization of human stem cell compartment and have revealed that cells with the same phenotype could be more heterogeneous than previously imagined. However, given the fact that the expression of lineage-priming genes does not necessarily imply that a cell would follow 1 single cell fate, functional validations are still required. Moreover, increasing the resolution of scRNAseq up to low-transcribed genes and microRNA would allow a better understanding of the regulatory programs controlling cell specification.
Even though performed on a very low number of cells and donor samples, somatic mutation detection proved to be a valid approach to study hematopoietic hierarchy in human subjects. The generation of tools that allow high-throughput analyses of thousands of single cells without the need for cell culture will be instrumental in making this strategy more quantitative and statistically valid for definitively unveiling in vivo dynamics, trafficking, and aging of the human hematopoietic system. mtDNA sequence variations provided the first promising approach for high-throughput unsupervised tracing of cell clones at single-cell resolution. However, the mitochondria capability of being transferred among cells could limit the applicability of this strategy.
Finally, clonal tracking studies performed in nonhuman primates and GT clinical trials allowed the characterization of the distinct role of HSPC subpopulations after transplantation, suggesting that efficacious transplantation protocols should preserve both primitive and committed progenitors in order to maintain initial hematopoietic reconstitution driven by highly proliferating progenitors, preventing rapid exhaustion of primitive HSCs, and to achieve LT graft maintenance. Given the fact that MPB HSPCs are becoming the preferred source for patients undergoing allogeneic transplantation and GT, assessing the dynamics of hematopoietic reconstitution and the capability of MPB to maintain gene correction in the long term would be of paramount importance. Indeed, gene expression and phenotypic profiling of BM and MPB CD34+ cells suggest that HSPCs have a different composition and cell state,69,70 which may result in different reconstitution kinetics and LT graft maintenance. In addition, after reaching the steady-state condition, posttransplant models could also be exploited to answer questions related to unperturbed hematopoiesis, confirming data from different approaches. Finally, monitoring barcoded hematopoiesis in LT GT patients reaching adulthood would generate novel insights on the aging hematopoietic system.
Overall, key questions on the “real-life” behavior and fate of HSPC in humans have now been answered, yet many more remain to be clarified. The integration of genomic, transcriptomic, and functional data with mathematical modeling has advanced our understanding of the mechanisms controlling HSPC self-renewal, differentiation, and aging in humans, and this information will expand the clinical application of HSPCs for the treatment of a wider array of diseases.
Acknowledgments
The authors thank Sophie Bevan for revising the manuscript.
This work was supported by Fondazione Telethon (TIGET Core Grant B2), the Italian Ministero della Salute (Programma di rete, NET-2011-02350069), and the European Commission (ERARE-3-JTC 2015 EUROCID).
Authorship
Contribution: S.S. and A.A. wrote the manuscript.
Conflict-of-interest disclosure: A.A. is the principal investigator of GT clinical trials for Wiskott Aldrich Syndrome, Severe Combined Immunodeficiency due to Adenosine Deaminase Deficiency, Metachromatic Leukodystrophy, and β-thalassemia sponsored by Orchard Therapeutics. S.S. declares no competing financial interests.
Correspondence: Alessandro Aiuti, San Raffaele Telethon Institute for Gene Therapy, Ospedale San Raffaele, Via Olgettina 58, 20132 Milan, Italy; e-mail: aiuti.alessandro@hsr.it.

