

Key Points
A distinct gene expression profile can predict EMR and long-term outcomes in CML.

Abstract
In chronic-phase chronic myeloid leukemia (CP-CML) patients treated with frontline imatinib, failure to achieve early molecular response (EMR; EMR failure: BCR-ABL1 >10% on the international scale at 3 months) is predictive of inferior outcomes. Identifying patients at high-risk of EMR failure at diagnosis provides an opportunity to intensify frontline therapy and potentially avoid EMR failure. We studied blood samples from 96 CP-CML patients at diagnosis and identified 365 genes that were aberrantly expressed in 13 patients who subsequently failed to achieve EMR, with a gene signature significantly enriched for stem cell phenotype (eg, Myc, β-catenin, Hoxa9/Meis1), cell cycle, and reduced immune response pathways. We selected a 17-gene panel to predict EMR failure and validated this signature on an independent patient cohort. Patients classified as high risk with our gene expression signature (HR-GES) exhibited significantly higher rates of EMR failure compared with low-risk (LR-GES) patients (78% vs 5%; P < .0001), with an overall accuracy of 93%. Furthermore, HR-GES patients who received frontline nilotinib had a relatively low rate of EMR failure (10%). However, HR-GES patients still had inferior deep molecular response achievement rate by 24 months compared with LR-GES patients. This novel multigene signature may be useful for selecting patients at high risk of EMR failure on standard therapy who may benefit from trials of more potent kinase inhibitors or other experimental approaches.
Introduction
Imatinib is the standard frontline treatment for patients with chronic-phase chronic myeloid leukemia (CP-CML), with up to 70% of patients achieving major molecular response (MMR).1 Of the remainder, some will progress to advanced-phase CML, some will be refractory to subsequent lines of therapy, and others may achieve good responses to salvage therapy with a more potent tyrosine kinase inhibitor (TKI).2-4 Failure to achieve early MR (EMR), defined as BCR-ABL1 value >10% on the international scale (IS) at 3 months, is predictive for inferior overall survival, progression-free survival, event-free survival (EFS), and failure-free survival (FFS).5-11 Additionally, rapid decrease in BCR-ABL1 transcripts, expressed as halving time7 study or log reduction,12 does have significant prognostic value. However, such information is only available at 3 months, when it may already be too late to intervene in some patients, because ∼50% of the patients who progress to blast crisis (BC) after EMR failure will do so within the first 12 months of therapy.6,11 This is a key rationale for further improving response prediction at the time of diagnosis.
Three baseline prognostic scoring systems, the Sokal,13 Hasford (Euro),14 and European Treatment and Outcome Study15 risk scores, have all been used to identify patients with a poor response and/or adverse prognosis in CP-CML.3,16,17 Recently, the European Treatment and Outcome Study long-term survival score (ELTS) was shown to be a strong predictor of overall survival in CML patients.18 However, these scores, by themselves, do not provide sufficient information for the prediction of achievement of early molecular targets.
Several gene expression profiling (GEP) studies have been reported to discriminate imatinib responders from nonresponders based on achievement of complete or partial cytogenetic response within 12 months of therapy.19-25 This study aimed to identify CP-CML patients who are at high risk of EMR failure and adverse clinical outcomes based on a gene expression signature (GES) assessed at diagnosis. Using this gene signature may inform therapeutic interventions at early time points, before treatment failure, potentially leading to improved clinical outcomes.
Materials and methods
Patient samples
This study was conducted according to the Declaration of Helsinki and approved by all appropriate ethics committees, with written informed consent obtained from all patients. Blood samples for the main study were sourced from patients enrolled in the TIDEL-II trial, with full details published elsewhere.11 Briefly, CP-CML patients were started on 600 mg of imatinib per day. Failure to achieve time-dependent molecular milestones (synonymous with optimal targets in 2013 by the European LeukemiaNet) led to either an increase in imatinib dose or a switch to nilotinib.11 Fresh mononuclear cells (MNCs) were isolated from peripheral blood (PB) collected at diagnosis using density gradient centrifugation.26 The PBMNCs were then lysed in TRIzol reagent (Invitrogen, Carlsbad, CA). Samples were available from 184 TIDEL-II patients, 96 of whom were randomly selected as the discovery cohort, whereas study results and outcome information from the remaining 88 patients were quarantined as an independent validation cohort. There were no significant differences with respect to baseline risk factors or EMR rate between the training and validation cohorts (Table 1).
Patient characteristics for discovery and validation cohorts
| Variable | Discovery cohort (n = 96) | Validation cohort (n = 88) | P |
|---|---|---|---|
| Age, y | .669 | ||
| Median | 48 | 50.5 | |
| Range | 17-77 | 17-80 | |
| Sex | .552 | ||
| Female | 38 (39.6) | 39 (44.3) | |
| Male | 58 (60.4) | 49 (55.7) | |
| BCR-ABL1 at 3 mo, % | 1 | ||
| <10 | 83 (86.5) | 77 (87.5) | |
| >10 | 13 (13.5) | 11 (12.5) | |
| BCR-ABL1 transcript type | .084 | ||
| b2a2 | 38 (39.6) | 36 (40.9) | |
| b2a3 | 1 (1.0) | 0 (0.0) | |
| b3a2 | 32 (33.3) | 39 (44.3) | |
| Both | 25 (26.0) | 12 (13.6) | |
| e1a2 | 0 (0.0) | 1 (1.1) | |
| Sokal score | .458 | ||
| High | 15 (16.9) | 19 (21.8) | |
| Intermediate | 27 (30.3) | 30 (34.5) | |
| Low | 47 (52.8) | 38 (43.7) | |
| ELTS | .442 | ||
| High | 7 (7.9) | 12 (13.8) | |
| Intermediate | 23 (25.8) | 20 (23.0) | |
| Low | 59 (66.3) | 55 (63.2) | |
| BC progression | .447 | ||
| Yes | 5 (5.2) | 2 (2.3) | |
| No | 91 (94.8) | 86 (97.7) | |
| MMR by 24 mo | .279 | ||
| Yes | 79 (82.3) | 66 (75.0) | |
| No | 17 (17.7) | 22 (25.0) | |
| MR4.5 by 5 y | .298 | ||
| Yes | 59 (61.5) | 47 (53.4) | |
| No | 37 (38.5) | 41 (46.6) |
| Variable | Discovery cohort (n = 96) | Validation cohort (n = 88) | P |
|---|---|---|---|
| Age, y | .669 | ||
| Median | 48 | 50.5 | |
| Range | 17-77 | 17-80 | |
| Sex | .552 | ||
| Female | 38 (39.6) | 39 (44.3) | |
| Male | 58 (60.4) | 49 (55.7) | |
| BCR-ABL1 at 3 mo, % | 1 | ||
| <10 | 83 (86.5) | 77 (87.5) | |
| >10 | 13 (13.5) | 11 (12.5) | |
| BCR-ABL1 transcript type | .084 | ||
| b2a2 | 38 (39.6) | 36 (40.9) | |
| b2a3 | 1 (1.0) | 0 (0.0) | |
| b3a2 | 32 (33.3) | 39 (44.3) | |
| Both | 25 (26.0) | 12 (13.6) | |
| e1a2 | 0 (0.0) | 1 (1.1) | |
| Sokal score | .458 | ||
| High | 15 (16.9) | 19 (21.8) | |
| Intermediate | 27 (30.3) | 30 (34.5) | |
| Low | 47 (52.8) | 38 (43.7) | |
| ELTS | .442 | ||
| High | 7 (7.9) | 12 (13.8) | |
| Intermediate | 23 (25.8) | 20 (23.0) | |
| Low | 59 (66.3) | 55 (63.2) | |
| BC progression | .447 | ||
| Yes | 5 (5.2) | 2 (2.3) | |
| No | 91 (94.8) | 86 (97.7) | |
| MMR by 24 mo | .279 | ||
| Yes | 79 (82.3) | 66 (75.0) | |
| No | 17 (17.7) | 22 (25.0) | |
| MR4.5 by 5 y | .298 | ||
| Yes | 59 (61.5) | 47 (53.4) | |
| No | 37 (38.5) | 41 (46.6) |
Data are n (%) unless otherwise noted.
An additional cohort of CP-CML patients, treated with nilotinib upfront through 2 prospective single-arm clinical studies (ENESTxtnd and PINNACLE), was included for comparison purposes. Both studies enrolled treatment-naïve CP-CML patients, who were then treated with 300 mg of nilotinib twice per day upfront until the 3-month time point, when EMR was determined.27,28
GEP microarray analysis
Genome-wide GEP was performed using the Illumina Human HT-12v4 platform (containing 47 323 probes) at the Australian Genome Research Facility (Melbourne, VIC, Australia). Microarray results were assessed for confounding batch effects using a multidimensional scaling plot (limma version 3.28.21),29 and raw intensities were normalized using the neqc function before inclusion for final analysis.30 Probes were filtered if not detected in any sample at P < .01. Pairwise comparisons were performed using empirical Bayes moderated t statistics (limma version 3.28.21).29 The false-discovery rate (FDR) was controlled using the Benjamini-Hochberg algorithm.31 Probes with FDR P < .05, and >1.5-fold were considered to be differentially expressed. The interactive gene expression plots can be assessed through shinyapps.io (https://chung-kok.shinyapps.io/CML_EMR_GEP/). Heatmap was generated by the pheatmap (version 1.0.8) Bioconductor package32 using Euclidean distance with Ward linkage parameters. All analyses were performed using R statistical software (version 3.3.2). The raw data can be assessed at National Center for Biotechnology Information Gene Expression Omnibus database (GSE130404).
GSEA
Gene set enrichment analysis (GSEA; version 2.2.4) was performed on a list of genes ranked by the moderated t statistics. GSEA was performed against the c2-curated gene sets from the Molecular Signatures Database (version 6.2; Broad Institute).33 Only gene sets with FDR q < 0.05 were considered statistically significant.
qRT-PCR (TaqMan Low Density Arrays) and bioinformatic analysis
Gene expression by quantitative reverse transcription polymerase chain reaction (qRT-PCR) was determined using the custom TaqMan Low Density Arrays (TLDA; Applied Biosystems). PBMNCs were the cell source for both imatinib- and nilotinib-treated cases. Total RNA was extracted from 1 × 106 to 1 × 107 MNCs using either TRIzol reagent or the miRNeasy RNA Extraction Kit (Qiagen, Chadstone Centre, VIC, Australia) and reverse transcribed to complementary DNA using the QuantiTect Reverse Transcription Kit (Qiagen) before being loaded onto the TLDA card. Results were analyzed with the QuantStudio 7 (Life Technologies) instrument. The raw data were normalized against endogenous control gene GUSB (Hs99999908_m1) using the delta Ct (ΔCt) method as implemented in the HTqPCR (version 1.26.0)34 Bioconductor package. Duplicate gene expression values were averaged for each gene for each sample before subsequent analyses.
For the construction of the multigene predictive model based on TLDA data, an ensemble gradient boosting tree method (xgboost; version 0.6-4)35 with synthetic minority oversampling technique36,37 was applied to the discovery cohort using fivefold cross-validation. The final model was chosen based on the minimum cross-validated classification errors using the discovery cohort. Xgboost is a direct application of gradient boosting for decision trees.35 Performance of the final model was assessed on an independent validation cohort. All analyses were performed using R statistical software (version 3.3.2). Additional information is provided in the supplemental Methods.
Statistical analyses for clinical end points
MR rates were calculated by cumulative incidence and compared using the Fine and Gray model implemented in the R package cmprsk (version 2.2-7) as previously described.7,38,39 An event was defined as achievement of the MR of interest, either MMR (BCR-ABL1 ≤0.1% on the IS) or MR4.5 (≤0.0032% [IS]) at 2 consecutive time points). Competing risks included permanent discontinuation of TIDEL-II treatment for any reason (including death).11
Survival analyses including EFS and FFS were performed using the Kaplan-Meier method, and comparisons were made using the log-rank test. An event in EFS was defined as progression to accelerated phase or BC, loss of either MMR or complete cytogenetic response, acquisition of BCR-ABL1 mutations, death, or European LeukemiaNet 2013 failure (BCR-ABL1 >10% [IS] at 6 months or BCR-ABL1 >1% [IS] at 12 months). FFS included all the EFS events as well as discontinuation of imatinib or nilotinib for any reason. Two-group comparison was performed using the Mann-Whitney U test for continuous variables and Fisher’s exact test for categorical variables. Analysis and graphical plots were performed using GraphPad Prism 7.0 (GraphPad Software Inc., La Jolla CA) or R statistical software (version 3.3.2).
Results
In our discovery cohort of 96 patients, 13 (14%) failed to achieve EMR (Table 1). Of the conventional prognostic markers, Sokal (P = .009) and ELTS (P = .005) were statistically significantly different between patients who achieved EMR vs those who did not (supplemental Table 1). As expected, patients who failed to achieve EMR had inferior rates of MMR by 24 months, compared with patients who achieved EMR (15% vs 93%; P < .0001; supplemental Figure 1A). Notably, of the patients who failed to achieve EMR, none achieved deep MR (MR4.5) by 5 years, compared with 76% of patients who achieved EMR (P < .0001; supplemental Figure 1B).
Next, we studied gene expression of patients who achieved EMR vs those who did not, performing pairwise comparisons using the Bioconductor R package limma29 (Figure 1A). There were 502 probes (corresponding to 365 annotated genes) identified as significantly differentially expressed (Figure 1B). In patients who failed to achieve EMR, 182 genes had decreased expression and 183 had increased expression, compared with those who achieved EMR (Figure 1B-C; supplemental Table 2). There seemed to be 4 distinct clusters of GEP for patients who achieved EMR. However, the clinical features and response achievement did not differ significantly between these groups (data not shown).
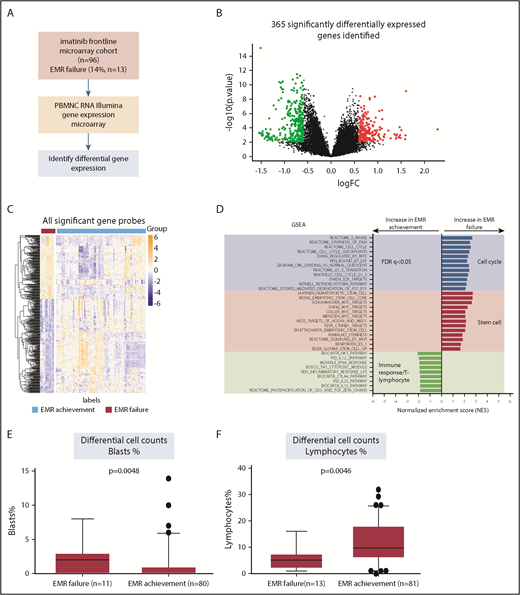
Distinct gene expression profile and enrichment of stem cells signature identified in diagnostic MNCs from patients who failed to achieve EMR. (A) The workflow of our study with regard to the discovery process, including samples from patients with EMR failure (n = 13) vs EMR achievement (n = 83), as defined by BCR-ABL1 percentage at 3 months. (B) Volcano plot demonstrating the effect of log2 fold change (FC) on the x-axis vs −log10 P value on the y-axis. Red circles indicate significant genes (FDR P < .05 and log2 FC > 0.6) with increased gene expression in the EMR failure patient group. Green circles indicate significant genes (FDR P < .05 and log2 FC < −0.6) with decreased gene expression in the EMR failure patient group. (C) Heatmap demonstrating the distinct gene expression patterns based on all significant probes (n = 502; FDR P < .05 and log2 FC > |0.6|). Orange represents increased gene expression level, and blue represents decreased gene expression level. The heatmap was generated using the pheatmap package. (D) GSEA indicates enrichment of stem cell signaling, cell cycle, and immune response/T lymphocytes in the EMR failure patient samples (BCR-ABL1 >10% IS at 3 months) compared with the EMR achievement patient samples (BCR-ABL1 ≤10% IS at 3 months). Blue bars represent cell cycle–related data sets. Red bars represent stem cell–related data sets. Green bars represent immune response/T lymphocyte–related signatures. Regarding normalized enrichment score (NES), positive score indicates positive enrichment in samples from patients who failed to achieve EMR, and negative score indicates enrichment in samples from patients who achieved EMR. (E) Boxplot displaying the differential blast percentage counts at diagnosis, indicating a significantly higher percentage in the samples collected from patients who failed to achieve EMR. Only 91 patients had differential blast percentage counts at diagnosis information available for analysis. (F) Boxplot displaying the lymphocyte percentage counts at diagnosis, indicating a significantly lower percentage in the EMR failure patient sample group. Only 94 patients had lymphocyte percentage counts at diagnosis information available for analysis. Statistical analysis was performed using the Mann-Whitney U test.
Distinct gene expression profile and enrichment of stem cells signature identified in diagnostic MNCs from patients who failed to achieve EMR. (A) The workflow of our study with regard to the discovery process, including samples from patients with EMR failure (n = 13) vs EMR achievement (n = 83), as defined by BCR-ABL1 percentage at 3 months. (B) Volcano plot demonstrating the effect of log2 fold change (FC) on the x-axis vs −log10 P value on the y-axis. Red circles indicate significant genes (FDR P < .05 and log2 FC > 0.6) with increased gene expression in the EMR failure patient group. Green circles indicate significant genes (FDR P < .05 and log2 FC < −0.6) with decreased gene expression in the EMR failure patient group. (C) Heatmap demonstrating the distinct gene expression patterns based on all significant probes (n = 502; FDR P < .05 and log2 FC > |0.6|). Orange represents increased gene expression level, and blue represents decreased gene expression level. The heatmap was generated using the pheatmap package. (D) GSEA indicates enrichment of stem cell signaling, cell cycle, and immune response/T lymphocytes in the EMR failure patient samples (BCR-ABL1 >10% IS at 3 months) compared with the EMR achievement patient samples (BCR-ABL1 ≤10% IS at 3 months). Blue bars represent cell cycle–related data sets. Red bars represent stem cell–related data sets. Green bars represent immune response/T lymphocyte–related signatures. Regarding normalized enrichment score (NES), positive score indicates positive enrichment in samples from patients who failed to achieve EMR, and negative score indicates enrichment in samples from patients who achieved EMR. (E) Boxplot displaying the differential blast percentage counts at diagnosis, indicating a significantly higher percentage in the samples collected from patients who failed to achieve EMR. Only 91 patients had differential blast percentage counts at diagnosis information available for analysis. (F) Boxplot displaying the lymphocyte percentage counts at diagnosis, indicating a significantly lower percentage in the EMR failure patient sample group. Only 94 patients had lymphocyte percentage counts at diagnosis information available for analysis. Statistical analysis was performed using the Mann-Whitney U test.
EMR failure is associated with a signature enriched for genes associated with stem cell signaling and a reduced number of T cell–associated genes
To gain biological insight into the significant genes associated with EMR failure, GSEA was performed. The differentially expressed genes associated with EMR failure were significantly enriched (FDR q < 0.05) for those associated with cell cycle and stem cell signaling/stemness (eg, Hoxa9/Meis1, Myc, β-catenin; Figure 1D). In contrast, the signature associated with the achievement of EMR was significantly enriched (FDR q < 0.05) for genes associated with immune response. On the basis of this association with stem cell signaling and immune response, differential cell counts were used to further interrogate the diagnostic PBMNC samples from patients who achieved or failed to achieve EMR. Patients with EMR failure had a significantly higher percentage of blast cells (P = .005; Figure 1E) and lower percentage of lymphocytes (P = .007; Figure 1F) at diagnosis, compared with patients who achieved EMR. This result was consistent with the results obtained from GSEA.
Prognostic significance of the gene expression–based classification
To develop a practical and cost-effective predictive assay to customize therapy for newly diagnosed CP-CML patients, we reduced our candidate gene set by selecting the most differentially expressed genes. This resulted in a 21-gene set, selected based on FDR P value and/or fold-change ranking (supplemental Table 3). Of these 21 genes, 3 had increased expression (IGFBP2, PRSS57, and CPXM1) in the EMR failure patient group compared with the EMR achievement patient group (supplemental Tables 2 and 4). Expression of these genes was confirmed via qRT-PCR on TLDA and visualized as a heatmap in our 96-patient discovery cohort (supplemental Figure 2). Using the extreme gradient boosting trees (xgboost)35 algorithm, a predictive model was constructed and refined to a binary classification model that predicted risk of EMR failure based on expression levels of 17 genes (Figure 2A; supplemental Figure 3A). The final model selected 17 genes as a minimum number of genes that had the lowest misclassification errors. In this model, IGFBP2 gene expression had the highest score, suggesting it was among the most important genes for predicting risk of EMR failure in the discovery cohort (Figure 2A). The output of the GES predictive model is a risk score for EMR failure ranging from 0 to 1. The default cutoff at 0.5 placed patients at high risk of EMR failure (>0.5) vs low risk of EMR failure (≤0.5).
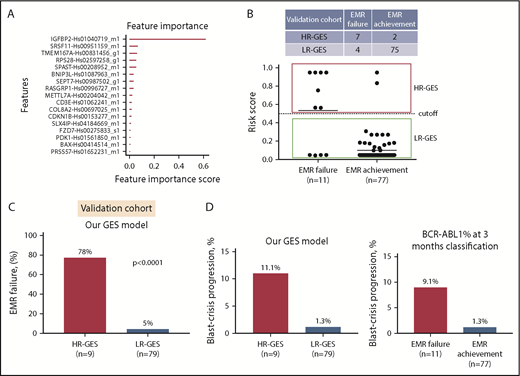
Prognostic significance of gene expression–based classification. (A) Bar plot displays the genes that are important contributors to the EMR failure prediction model. The classifier parameters were determined by fivefold cross-validation, yielding a binary classification model that predicts risk of EMR failure based on expression levels of 17 genes. (B) Scatter plot demonstrates the risk score by our predictive model for all 88 patients (EMR achievement or EMR failure) in the independent validation cohort. By default, high risk of EMR failure (HR-GES) was defined as risk score >0.5, and low risk of EMR failure (LR-GES) was defined as risk score ≤0.5. According to this model, 9 (10%) and 79 patients (90%) were classified as HR-GES and LR-GES, respectively. (C) Bar plot of patient samples assigned as HR-GES (n = 9), where 78% failed to achieve EMR compared with 5% of patient samples assigned as LR-GES (n = 79) in the independent validation cohort. Statistical analysis was performed using Fisher’s exact test. (D) Bar plot demonstrating high-risk patient group classified by our predictive model (left) and EMR failure patient group (classified by BCR-ABL1 percentage at 3 months; right). HR-GES indicates a higher BC progression rate compared with low-risk patient group classified by our model or EMR achievement patient group (classified by BCR-ABL1 level at 3 months), respectively.
Prognostic significance of gene expression–based classification. (A) Bar plot displays the genes that are important contributors to the EMR failure prediction model. The classifier parameters were determined by fivefold cross-validation, yielding a binary classification model that predicts risk of EMR failure based on expression levels of 17 genes. (B) Scatter plot demonstrates the risk score by our predictive model for all 88 patients (EMR achievement or EMR failure) in the independent validation cohort. By default, high risk of EMR failure (HR-GES) was defined as risk score >0.5, and low risk of EMR failure (LR-GES) was defined as risk score ≤0.5. According to this model, 9 (10%) and 79 patients (90%) were classified as HR-GES and LR-GES, respectively. (C) Bar plot of patient samples assigned as HR-GES (n = 9), where 78% failed to achieve EMR compared with 5% of patient samples assigned as LR-GES (n = 79) in the independent validation cohort. Statistical analysis was performed using Fisher’s exact test. (D) Bar plot demonstrating high-risk patient group classified by our predictive model (left) and EMR failure patient group (classified by BCR-ABL1 percentage at 3 months; right). HR-GES indicates a higher BC progression rate compared with low-risk patient group classified by our model or EMR achievement patient group (classified by BCR-ABL1 level at 3 months), respectively.
To examine our predictive model performance, the final classification model was then applied to the independent validation cohort (n = 88). In the validation cohort, 9 (10%) and 79 patients (90%) were classified as HR-GES and LR-GES, respectively (Figure 2B). The sensitivity and specificity of this model were 64% and 97%, respectively (Figure 2B; supplemental Figure 3B), with an overall accuracy of 93% (95% confidence interval [CI:], 86%-98%). A total of 6 patients were misclassified based on our model (Table 2). Importantly, those patients assigned to the HR-GES group had a 78% risk of EMR failure, compared with only 5% in patients assigned to the LR-GES group (P < .0001; odds ratio, 65.6; 95% CI, 10.7-340.1; Figure 2C). There was no statistically significant association between our GES score and Sokal score (P = .35), ELTS (P = .29), cytogenetic abnormalities at baseline (P = 1.00), BCR-ABL1 transcript type (P = 1.00), or baseline BCR-ABL1 level (P = .17; supplemental Table 5). Although we observed differences in blast and lymphocyte counts when comparing EMR failure with EMR achievement patient groups, we further analyzed the relationship between blast count, lymphocyte count, spleen size, eosinophil count, basophil counts, platelet count, and outcome (specifically, EMR) in our entire cohort of 184 patients. Univariate analyses showed that lymphocyte count, spleen size, and GES score were statistically significantly associated with EMR outcome (supplemental Table 6). However, blast count was a minor component of the predictor for EMR (P = .12; supplemental Table 6). The strength of the relationships was preserved in a multivariate model including our GES score, lymphocyte count, spleen size, and blast count as covariates. We found the GES score offered better prediction of EMR (hazard ratio [HR], 0.028; P = 8.2e-08; supplemental Table 6) compared with lymphocyte count (HR, 1.02; P = .003; supplemental Table 6), whereas blast count (HR, 1.05; P = .22; supplemental Table 6) and spleen size (HR, 0.98; P = .19; supplemental Table 6) had only a weak relationship.
Detailed information about patients misclassified by our predictive model
| Patient ID | BCR-ABL1 % at diagnosis | BCR-ABL1 % at 3 mo | Sokal score | ELTS | BCR-ABL1 transcript type | TKIs | MMR by 12 mo | MR4.5 by 5 y | Withdrawal |
|---|---|---|---|---|---|---|---|---|---|
| EMR failure patients (n = 4) classified as low risk | |||||||||
| 145* | 55 | 14 | Int | Int | b2a2 | IM | N/A | N/A | Yes |
| 109 | 85 | 31 | High | High | b3a2 | IM, NIL | No | Yes | No |
| 102 | 124 | 13 | Low | Low | b3a2 | IM | Yes | No | No |
| 170 | 73 | 18 | Low | Low | b3a2 | IM, NIL | No | Yes | No |
| EMR achievement patients (n = 2) classified as high risk | |||||||||
| 183 | 89 | 1.50 | Int | Int | b2a2 | IM, NIL | No | No | No |
| 159 | 86 | 6.90 | High | High | b3a2 | IM, NIL | No | No | No |
| Patient ID | BCR-ABL1 % at diagnosis | BCR-ABL1 % at 3 mo | Sokal score | ELTS | BCR-ABL1 transcript type | TKIs | MMR by 12 mo | MR4.5 by 5 y | Withdrawal |
|---|---|---|---|---|---|---|---|---|---|
| EMR failure patients (n = 4) classified as low risk | |||||||||
| 145* | 55 | 14 | Int | Int | b2a2 | IM | N/A | N/A | Yes |
| 109 | 85 | 31 | High | High | b3a2 | IM, NIL | No | Yes | No |
| 102 | 124 | 13 | Low | Low | b3a2 | IM | Yes | No | No |
| 170 | 73 | 18 | Low | Low | b3a2 | IM, NIL | No | Yes | No |
| EMR achievement patients (n = 2) classified as high risk | |||||||||
| 183 | 89 | 1.50 | Int | Int | b2a2 | IM, NIL | No | No | No |
| 159 | 86 | 6.90 | High | High | b3a2 | IM, NIL | No | No | No |
IM, imatinib; int, intermediate; N/A, not applicable; NIL, nilotinib.
Withdrawn at 6 months because of poor patient compliance.
Progression status in the HR-GES patient group
EMR failure is significantly associated with progression to BC.5,7,11 Only 2 (2%) of 88 patients in our validation cohort progressed to BC. In the HR-GES patient group, a higher frequency (n = 1 [11%] of 9) of patients progressing to BC was observed, compared with 1% (n = 1 of 79) of patients in the LR-GES patient group (Figure 2D). The predictive power of the GES score demonstrated here is similar to prognostic information derived from BCR-ABL1 percentage at 3 months, although our small number of events limits the power of this analysis.
HR-GES patient group had inferior long-term MR and survival
We then investigated the association between our predictive model and longer-term outcomes, such as MMR at 24 months and MR4.5 by 60 months, and compared its performance against established markers, such as stratification using BCR-ABL1 percentage at 3 months. Using patients from the independent validation cohort alone, we found HR-GES patients had significantly lower achievement of MMR by 24 months compared with the LR-GES patient group (44% vs 78%; P = .04; Figure 3A). This is similar to the predictive value of BCR-ABL1 percentage at 3 months (Figure 3B). Furthermore, none of the HR-GES patients achieved deep MR (MR4.5) by 5 years compared with LR-GES patient group (0% vs 63%; P = .004; Figure 3C). As expected, BCR-ABL1 percentage achieved at 3 months also predicted deep MR achievement (30% vs 61%; P = .02; Figure 3D), although this metric seemed less discriminatory than our GES classification.

The high-risk patient group classified by our predictive model had inferior MRs compared with the low-risk patient group in the validation cohort. (A) The high-risk (HR-GES) patient group classified by our predictive model (n = 9) had significantly inferior cumulative incidence (44%) of MMRs (>3 log reduction of BCR-ABL1 transcript value) by 24 months compared with the low-risk (LR-GES) patient group (78%; n = 79). (B) The EMR failure patient group defined by BCR-ABL1 percentage at 3 months (n = 11) had significantly inferior cumulative incidence of MMRs (>3 log reduction of BCR-ABL1 transcript value) by 24 months (45%) compared with the EMR achievement patient group (79%; n = 77). (C) The high-risk patient group classified by our predictive model (n = 9) had significantly inferior cumulative incidence of deep MR (MR4.5; >4.5 log reduction of BCR-ABL1 transcript value) by 5 years (0%) when compared with the low-risk patient group (63%; n = 79). (D) The EMR failure patient group defined by BCR-ABL1 percentage at 3 months (n = 11) had significantly inferior cumulative incidence of MR4.5 by 5 years (30%) compared with the EMR achievement patient group (61%; n = 77). All statistical analyses were performed using the Fine and Gray test.
The high-risk patient group classified by our predictive model had inferior MRs compared with the low-risk patient group in the validation cohort. (A) The high-risk (HR-GES) patient group classified by our predictive model (n = 9) had significantly inferior cumulative incidence (44%) of MMRs (>3 log reduction of BCR-ABL1 transcript value) by 24 months compared with the low-risk (LR-GES) patient group (78%; n = 79). (B) The EMR failure patient group defined by BCR-ABL1 percentage at 3 months (n = 11) had significantly inferior cumulative incidence of MMRs (>3 log reduction of BCR-ABL1 transcript value) by 24 months (45%) compared with the EMR achievement patient group (79%; n = 77). (C) The high-risk patient group classified by our predictive model (n = 9) had significantly inferior cumulative incidence of deep MR (MR4.5; >4.5 log reduction of BCR-ABL1 transcript value) by 5 years (0%) when compared with the low-risk patient group (63%; n = 79). (D) The EMR failure patient group defined by BCR-ABL1 percentage at 3 months (n = 11) had significantly inferior cumulative incidence of MR4.5 by 5 years (30%) compared with the EMR achievement patient group (61%; n = 77). All statistical analyses were performed using the Fine and Gray test.
Patients with HR-GES had significantly inferior EFS compared with LR-GES patients (53% vs 83%; P = .019; HR, 3.5; 95% CI, 0.6-20.3; Figure 4A); again, this was similar to stratification by 3-month BCR-ABL1 value (59% vs 83%; P = .053; Figure 4B). A similar pattern emerged in analysis with respect to FFS (GES: 44% vs 72%; P = .025; HR, 2.9; 95% CI, 0.7-12.4 by GES; Figure 4C and 3-month BCR-ABL1 percentage: 45% vs 73%; P = .014; Figure 4D).

The high-risk patient group classified by our predictive model demonstrated inferior clinical outcomes compared with the low-risk patient group in the validation cohort. (A) The high-risk (HR-GES) patient group classified by our predictive model (n = 9) had significantly inferior EFS by 5 years (53%) compared with the low-risk (LR-GES) patient group (83%; n = 79). (B) The EMR failure patient group classified by BCR-ABL1 percentage at 3 months (n = 11) had inferior EFS by 5 years (59%) compared with the EMR achievement patient group (83%; n = 77). (C) The high-risk patient group classified by our predictive model (n = 9) had significantly inferior FFS by 5 years (44%) when compared with the low-risk patient group (72%; n = 79). (D) The EMR failure patient group classified by BCR-ABL1 percentage at 3 months (n = 11) had inferior FFS by 5 years (45%) when compared with the EMR achievement patient group (73%; n = 77). All statistical analyses were performed using the log-rank test.
The high-risk patient group classified by our predictive model demonstrated inferior clinical outcomes compared with the low-risk patient group in the validation cohort. (A) The high-risk (HR-GES) patient group classified by our predictive model (n = 9) had significantly inferior EFS by 5 years (53%) compared with the low-risk (LR-GES) patient group (83%; n = 79). (B) The EMR failure patient group classified by BCR-ABL1 percentage at 3 months (n = 11) had inferior EFS by 5 years (59%) compared with the EMR achievement patient group (83%; n = 77). (C) The high-risk patient group classified by our predictive model (n = 9) had significantly inferior FFS by 5 years (44%) when compared with the low-risk patient group (72%; n = 79). (D) The EMR failure patient group classified by BCR-ABL1 percentage at 3 months (n = 11) had inferior FFS by 5 years (45%) when compared with the EMR achievement patient group (73%; n = 77). All statistical analyses were performed using the log-rank test.
HR-GES patient group had a much lower risk of EMR failure if treated with nilotinib upfront
We then examined whether patients classified as having a high risk of EMR failure would have had a different trajectory if a more potent TKI had been chosen as frontline treatment. We applied our GES risk score in 132 CP-CML patients who were treated with nilotinib upfront from 2 prospective clinical studies.27,28 Nilotinib, a second-generation TKI, is significantly more potent than imatinib, resulting in a lower transformation rate and higher MR rate for patients treated in the frontline setting.6,27,40 The EMR failure rate in our nilotinib cohort was 3.8%. Of the 132 patients, 20 (15%) were classified as HR-GES and 112 (85%) as LR-GES. Interestingly, HR-GES patients had a much lower risk of EMR failure if they were treated with nilotinib upfront, compared HR-GES patients treated with imatinib upfront (10% vs 78%; 2-sample z test P < .001; Figure 5A). The risk of EMR failure for LR-GES patients was low regardless of whether they were treated with frontline nilotinib (3%) or imatinib (5%; 2-sample z test P = .388; Figure 5B). Next, we examined other response measures, such as MR4.5 and MMR, in HR-GES patients treated with nilotinib upfront when compared with LR-GES patients. Of the 132 patients, 75 had 24-month follow-up MR data. As with the analysis using TIDEL-II patients, no HR-GES patient achieved MR4.5 by 24 months, compared with 29% in the LR-GES group (P = .067; Figure 5C). The MMR achievement rate was uniformly high in both groups (P = .55; Figure 5D).

Our predicted HR-GES patient group had a much lower risk of EMR failure if treated with nilotinib upfront. (A) Bar plot demonstrates comparison of the EMR failure rate in patient samples assigned as high risk (HR-GES) by our predictive model if treated with imatinib (78%; n = 9) vs nilotinib upfront (10%; n = 20). (B) Bar plot demonstrates comparison of the EMR failure rate in patient samples assigned as low risk (LR-GES) by our predictive model if treated with imatinib (5%; n = 79) or nilotinib upfront (3%; n = 112). (C) Of the 132 patients treated with nilotinib upfront, there were 75 patients who had 24-month follow-up MR data. The high-risk patient group classified by our predictive model (n = 9) had inferior cumulative incidence of deep MR (MR4.5; >4.5 log reduction of BCR-ABL1 transcript value) by 24 months (0%) when compared with the low-risk patient group (29%; n = 66; P = .067). Statistical analysis was performed using the Fine and Gray test. (D) The high-risk patient group classified by our predictive model (n = 9) had no statistically significant difference in cumulative incidence of MMRs (>3 log reduction of BCR-ABL1 transcript value) by 24 months (78%) compared with the low-risk patient group (85%; n = 66; P = .55). Statistical analysis was performed using the Fine and Gray test.
Our predicted HR-GES patient group had a much lower risk of EMR failure if treated with nilotinib upfront. (A) Bar plot demonstrates comparison of the EMR failure rate in patient samples assigned as high risk (HR-GES) by our predictive model if treated with imatinib (78%; n = 9) vs nilotinib upfront (10%; n = 20). (B) Bar plot demonstrates comparison of the EMR failure rate in patient samples assigned as low risk (LR-GES) by our predictive model if treated with imatinib (5%; n = 79) or nilotinib upfront (3%; n = 112). (C) Of the 132 patients treated with nilotinib upfront, there were 75 patients who had 24-month follow-up MR data. The high-risk patient group classified by our predictive model (n = 9) had inferior cumulative incidence of deep MR (MR4.5; >4.5 log reduction of BCR-ABL1 transcript value) by 24 months (0%) when compared with the low-risk patient group (29%; n = 66; P = .067). Statistical analysis was performed using the Fine and Gray test. (D) The high-risk patient group classified by our predictive model (n = 9) had no statistically significant difference in cumulative incidence of MMRs (>3 log reduction of BCR-ABL1 transcript value) by 24 months (78%) compared with the low-risk patient group (85%; n = 66; P = .55). Statistical analysis was performed using the Fine and Gray test.
Discussion
Here we show the development and validation of a tool to provide additional information for prognostication at the time of diagnosis to complement currently available predictive markers. Patients classified as having a low risk of EMR failure may then be given imatinib, a drug with an excellent long-term safety record. Patients with a high risk of EMR failure can be identified at diagnosis and potentially offered a more potent TKI, if their comorbidity profile is acceptable. We speculate that this high-risk group could benefit further from the addition of novel interventional strategies, such as the addition of pegylated interferon or asciminib.41-43
GEP has been used previously to develop prognostic tools, in many different cancer settings.44-49 In CML, several gene signatures have used different cell fractions (eg, CD34s, MNCs, and total white cells, from either PB or bone marrow) and various algorithms for analysis.19-25 Importantly, none thus far has been developed to predict EMR.
A number of risk stratification strategies are currently available and already in clinical use. Clinical scoring systems, such as Sokal score, Hasford score, and the more recent ELTS and algorithms combining their components, can be implemented at diagnosis, but they have limited sensitivity and specificity for the prediction of achievement of early molecular targets. Cytogenetic analysis at diagnosis, a key component of risk stratification, only identifies a minor subset of patients at higher risk of treatment failure.50 Genomic screening for somatic variants may provide further prognostic information, although currently outcome data and expertise in genomic screening application are limited.51 The level of BCR-ABL1 at 3 months, or EMR, has excellent prognostic ability, although interventions based on this metric may already be too late for effective salvage in many patients.
This study demonstrates that a multigene expression signature using qRT-PCR can identify a group of patients at the time of diagnosis who have a very high risk of subsequent EMR failure as well as inferior long-term survival and molecular outcomes. The model provides prognostic information beyond EMR; patients who were classified in the high-risk group by our GES model had inferior long-term MRs even if they achieved EMR by 3 months. We also found that upfront therapy with nilotinib, a more potent TKI, greatly lowered the risk of EMR failure in the HR-GES cohort (78% EMR failure with imatinib vs 10% with nilotinib). However, these patients remain at high risk of inferior long-term outcomes as compared with their peers in the same cohort, as judged by the inferior rate of MR4.5 achievement at 24 months. This suggests that a more potent TKI alone may not be sufficient to completely overcome the negative prognostic impact of an HR-GES score.
In contrast to HR-GES patients, the LR-GES patient group had a low EMR failure rate (3% to 5%) regardless of whether imatinib or nilotinib was used as frontline treatment. This suggests that imatinib, a drug widely available off patent in many countries and with a favorable safety profile, may be used in the LR-GES patient group, leading to a very low risk of EMR failure and adverse outcomes.
We envisage that in the future, other prognostic biomarkers will be combined with our GES score to further improve our capacity to pursue a risk-adapted approach to frontline therapy. In developing predictors of response in CP-CML based on intrinsic features of the disease at diagnosis, we and others recognize that many cases of treatment failure may be unrelated to the intrinsic biology of the disease. Rather, factors such as poor drug adherence, inadequate drug levels (because of pharmacokinetic factors), and treatment interruptions are known to significantly affect outcome. These factors cannot be adequately modeled from a disease biology perspective, and a margin of error is to be expected from all prognostic algorithms.
In our study, we used MNCs as the starting material for GEP. We recognize that this may be a departure from usual laboratory practices, where RNA derived from total white cells, rather than MNCs, is more widely available. However, the isolation of MNCs is not technically challenging and adds minimally to the workflow in a diagnostic laboratory, and MNCs have traditionally been preferred as the starting material for CML biological investigations, because they may better reflect the cellular populations that determine CML biology.
In addition to its utility as a clinical aid, our gene signature may also provide insights into the underlying disease biology of resistant phenotypes. Among the 17 genes, we found 8 (IGFBP2, SRSF11, BAX, CDKN1B, BNIP3L, FZD7, PRSS57, and RPS28) that overlapped with other TKI resistance and CML progression studies, including the acute myeloid leukemia LSC17 study.20,21,23,52-56 Particularly, IGFBP2 and PRSS57, which were expressed significantly higher in the EMR failure group compared with the EMR achievement group, were found to be associated with stem cell signaling.54,57-59 Additionally, high IGFBP2 expression has been associated with imatinib resistance in CML patients, dasatinib resistance in lung cancer, and tumor cell survival and chemotherapy resistance in AML.52,59-62 Interestingly, IGFBP2 stabilizes β-catenin and regulates its nuclear functions through inactivation of GSK3β.57 This suggests that these genes may play important biological roles in regulating leukemic stem cells associated with more aggressive disease. The biological significance of these genes and their roles in promoting treatment resistance comprise an area of future study with potentially broader significance for leukemia.
In summary, we were able to risk stratify newly diagnosed CP-CML patients receiving frontline imatinib using a multigene expression signature derived from diagnostic MNCs isolated from a PB sample. Further confirmation of the predictive performance of this model, using a large independent patient cohort, is now indicated. This should include not only imatinib-treated patients, but also patients treated with frontline second-generation TKIs. We further postulate that this prognostic gene signature could be used in combination with other known risk factors in a composite risk score, which would likely include clinical risk features as identified in the ELTS, for recognizing high-risk patients who would benefit from personalized therapeutic approaches to obtain optimal clinical outcomes.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge Kelly Quek, who provided advice on bioinformatic analysis, and Jennifer McLean, who provided technical assistance.
The scientific work was supported by National Health and Medical Research Council (NHMRC) funding with material from patients who participated in the TIDEL-II study, which was supported in part by grants from Novartis Pharmaceuticals Australia. This study was sponsored by the Australasian Leukaemia and Lymphoma Group; undertaken with the financial support of Cancer Council SA’s Beat Cancer Project on behalf of its donors and the State Government through the Department of Health; and supported by a Translational Research Program from The Leukemia & Lymphoma Society. D.T.Y. is an NHMRC Early Career Research Fellow. T.P.H. is an NHMRC Principal Research Fellow.
Novartis had no role in gathering, analyzing, or interpreting the data.
Authorship
Contribution: C.H.K. conducted the study, performed TLDA, bioinformatic, and statistical analyses, gathered and analyzed data, wrote the first draft of the manuscript, and designed the study; D.T.Y. conducted the TIDEL-II study, contributed patient samples, gathered and analyzed the data, performed statistical analysis, and critically revised the manuscript; L.L. conducted the study, analyzed the data, and contributed to the manuscript; D.B.W. performed experiments and contributed to the manuscript; T.M.L. performed TLDA and contributed to the manuscript; P.D. conducted the study, performed experiments, and contributed to the manuscript; V.A.S. collected and processed the samples, designed the study, and contributed to the manuscript; J.R. designed the TIDEL-II study, analyzed the data, and contributed to the manuscript; D.L.W. designed the study, gathered and analyzed data, and contributed to the manuscript; and T.P.H. conducted the TIDEL-II, ENESTxtnd, and PINNACLE studies, designed the correlative studies, supervised the conduct of the study, contributed patient samples, served on the study management committee, and contributed to the manuscript.
Conflict-of-interest disclosure: D.T.Y. receives research funding from Novartis and Bristol-Myers Squibb and receives honoraria from and participates in advisory boards of Ariad, Novartis, Amgen, Pfizer, and Bristol-Myers Squibb. J.R. is a former employee of Novartis AG and holds stock in the company. D.L.W. receives research funding from Ariad, CSL Behring, Novartis, and Bristol-Myers Squibb and receives honoraria from and participates in advisory boards of Novartis and Bristol-Myers Squibb. T.P.H. receives funding from Ariad, CSL Behring, Novartis, and Bristol-Myers Squibb and receives honoraria from and participates in advisory boards of Ariad, Pfizer, Novartis, and Bristol-Myers Squibb. The remaining authors declare no competing financial interests.
Correspondence: Timothy P. Hughes, Precision Medicine Theme Leader, South Australian Health and Medical Research Institute, North Terrace, Adelaide, SA 5000, Australia; e-mail: tim.hughes@sahmri.com.

