

Key Points
Tranexamic acid reduces postsurgical infection rates.
Patients with diabetes are refractory to the effects of tranexamic acid.

Abstract
Tranexamic acid (TXA) is an antifibrinolytic agent that blocks plasmin formation. Because plasmin is known to promote inflammatory and immunosuppressive responses, we explored the possibility that plasmin-mediated immunosuppression in patients undergoing cardiac surgery can be directly reversed by TXA and decrease postoperative infection rates. The modulatory effect of TXA on inflammatory cytokine levels and on innate immune cell activation were evaluated with multiplex enzyme-linked immunosorbent assay and flow cytometry, respectively. Postoperative infection rates were determined in patients undergoing cardiac surgery and randomized to TXA (ACTRN12605000557639; http://www.anzca.edu.au). We demonstrate that TXA-mediated plasmin blockade modulates the immune system and reduces surgery-induced immunosuppression in patients following cardiac surgery. TXA enhanced the expression of immune-activating markers while reducing the expression of immunosuppressive markers on multiple myeloid and lymphoid cell populations in peripheral blood. TXA administration significantly reduced postoperative infection rates, despite the fact that patients were being administered prophylactic antibiotics. This effect was independent of the effect of TXA at reducing blood loss. TXA was also shown to exert an immune-modulatory effect in healthy volunteers, further supporting the fibrin-independent effect of TXA on immune function and indicating that baseline plasmin levels contribute to the regulation of the immune system in the absence of any comorbidity or surgical trauma. Finally, the capacity of TXA to reduce infection rates, modulate the innate immune cell profile, and generate an antifibrinolytic effect overall was markedly reduced in patients with diabetes, demonstrating for the first time that the diabetic condition renders patients partially refractory to TXA.
Introduction
Uncontrolled bleeding and infection still remain significant risk factors in individuals undergoing extensive surgery. The most widely used antifibrinolytic agent to reduce bleeding is tranexamic acid (TXA).1 TXA is a lysine analog that blocks the binding of plasminogen to exposed lysine residues on fibrin,1 thereby shielding fibrin from proteolytic attack. Plasmin also plays important roles in areas unrelated to fibrinolysis.2 Plasmin(ogen) can act in a proinflammatory manner via triggering chemotaxis3,4 and cytokine release,5 but it is also involved in the resolution of inflammation.6 Moreover, plasmin can promote phagocytosis in macrophages and dendritic cells7-9 and inhibit dendritic cell migration to draining lymph nodes,8 with the latter implicating plasmin in the promotion of an immunosuppressive state. This immunosuppressive capacity of the fibrinolytic system has more recently been shown to occur in a mouse model of ischemic stroke following thrombolysis.10 Hence, we investigated the net effect of plasmin inhibition on the immune profile in patients undergoing cardiac surgery and in healthy volunteers.
Methods
Healthy volunteer recruitment
Volunteers (8 males and 2 females, mean age 35.6 years) agreed to take 1 g of TXA orally. A total of 2.7 mL of blood was drawn into a citrated vacutainer tube (BD Biosciences) before the intake of TXA, as well as 2, 4, and 24 hours thereafter, as approved by the Alfred Hospital Research Ethics Committee. All participants provided written informed consent.
ATACAS trial design
The Aspirin and Tranexamic Acid for Coronary Artery Surgery (ATACAS) trial (ACTRN12605000557639; http://www.anzca.edu.au) was a multicenter double-blind randomized controlled trial of TXA in patients undergoing coronary artery surgery and who were at increased risk for complications.11,12 Inclusion/exclusion criteria are provided in supplemental Table 1. The ATACAS trial and this substudy were approved by the Alfred Hospital Research Ethics Committee. Each participant provided written informed consent.
Patients
Patient enrollment occurred at our institution from March 2006 to October 2015. The trial flow diagram is presented in supplemental Figure 1. We prospectively enrolled 379 patients prior to commencing data collection for this study, and we were able to retrieve the medical records of 374 (98.7%) patients; an additional 239 patients were enrolled prospectively. Baseline demographic and clinical characteristics of patients were well balanced in the treatment groups (supplemental Table 2).
We characterized changes in cytokines and circulating cellular immune profile in 41 randomized cardiac surgery patients consecutively enrolled into the ATACAS trial, at the beginning of surgery (before administration of placebo/TXA), as well as on postoperative day 1 (POD-1) and POD-3.
Trial drug
TXA was administered IV after induction of anesthesia, but before cardiopulmonary bypass, at a dose of 100 mg/kg over 30 minutes for the first 183 patients at the lead site. This dose was reduced to 50 mg/kg for all subsequent patients because of a concern for seizure risk.12
Clinical management
All patients received routine surgical and other perioperative care, including antibiotic prophylaxis (supplemental Table 2), surgical hemostasis, and any use of blood transfusion. Most surgical site infections (SSIs) were not cultured but were treated empirically with gram-positive cover (typically cefazolin).
Trial end points
The primary end point was the rate of health care–associated infection in the first 30 days postoperatively. Definitions for each type of infection are presented in supplemental Table 3. Secondary outcomes were specific sites of infection, surgical blood loss, blood transfusions, and duration of intensive care and hospital stay. All patients had their medical records reviewed and were contacted at 30 days after surgery to identify trial end points and other adverse events. For patients who had documentation of an SSI only at the time of their 6-week clinic review, it was assumed that this infection was present within the 30-day postoperative period. All infection data collection was performed by research staff blinded to treatment group assignment.
Subgroups
Diabetes status was a prespecified subgroup. We compared differences in the primary outcome between patients with and without diabetes using a log-binomial regression model. In a post hoc analysis, we tested whether exposure to a blood transfusion led to TXA having a differential effect on the incidence of infection.
Sample size
The main ATACAS factorial trial required 4484 patients to detect a clinically important reduction (from 10% to 7%) in the primary outcome of death and thrombotic events.12 A sample size estimation was not made for this infection substudy; we collected infection data on all subsequently enrolled ATACAS patients at the lead site while still blinded to treatment.
Statistical analysis for clinical outcome
Dichotomous end points were analyzed using the χ2 test; results are expressed as risk ratios (RRs) with 95% confidence intervals (CIs). Generalized linear models were constructed from binomial regression with a logarithmic link to evaluate interactions. For the TXA comparisons, separate models were fitted for diabetes and exposure to blood transfusion. All reported P values are 2 sided and were not adjusted for multiple comparisons. P < .05 was considered statistically significant. We used Statistical Package for the Social Sciences (Version 23; SPSS Inc., Chicago, IL) for analysis.
Sample collection and laboratory analysis
The 41 consecutive patients of the study population were analyzed for immunological changes in blood and plasma; 19 were given placebo, and 22 were administered TXA. Baseline characteristics are shown in supplemental Table 4.
Blood was drawn into a 10-mL K2-EDTA tube and a 2.7-mL citrate vacutainer tube (BD Biosciences) before surgery and TXA administration (preoperative), at the end of surgery, and on POD-1 and POD-3.
Flow cytometry
Flow cytometric analysis of whole blood (200 μL within 3 hours of collection), as well as the gating strategy using 2 15-color antibody panels (supplemental Table 5) designed and optimized to characterize myeloid and lymphoid cell populations, has been described.13
Collection of plasma for cytokine analysis
Blood tubes were centrifuged at 800g for 15 minutes at 21°C. Plasma was transferred into Eppendorf tubes, which were recentrifuged at 16 000g for 2 minutes at 21°C to generate platelet-poor plasma. Plasma was then transferred into a new Eppendorf tube and stored at −80°C.
Cytokine multiplex enzyme-linked immunosorbent assay
A human cytokine antibody array kit, Human ProcartaPlex Panel (eBioscience), detecting 10 analytes (interleukin-6 [IL-6], interferon-γ [IFN-γ], tumor necrosis factor-α [TNF-α], soluble TNF receptor 2 [sTNFR2], IL-β, IL-13, IL-8, MCP-1, IL-10, and tumor growth factor-β [TGF-β]) on a Luminex platform, was used following the instructions provided. Samples were tested in duplicate.
Evaluation of plasma D-dimer levels
D-dimer levels were determined in citrated plasma samples from 11 placebo- and 13 TXA-treated patients using a latex agglutination assay performed by the Pathology Service of Alfred Hospital.
Statistical analysis of laboratory parameters
Statistical outliers were identified with a ROUT outlier test (Q = 1%); when an outlier was detected, the participant was removed from the comparison. For changes in immune parameters over time, a repeated-measures 1-way analysis of variance (ANOVA) with Dunnett’s correction was performed. Comparisons between placebo and TXA effects at individual time points were presented as fold change from preoperative values and evaluated using a 2-tailed Student t test. Results were considered significant with a P < .05.
Results
The effect of TXA on postoperative infection rates
We evaluated bleeding and infection outcomes in 613 patients recruited into the ATACAS trial.12 Details of the organisms grown are shown in supplemental Table 6. TXA treatment significantly reduced SSIs (P = .041) (Table 1), despite the fact that the placebo- and TXA-treated cohorts were administered prophylactic antibiotics (supplemental Table 2). The incidence of overall health care–associated infection in patients receiving TXA was also reduced, but not significantly (P = .059) (Table 1). Although the majority of infections were superficial SSIs, the point estimates of the RRs for sepsis, bacteremia, and pneumonia were all <1.0 (ie, lower risk). Indeed, the incidence of sepsis in the 2 groups was 12.1% vs 15.7% (RR, 0.87; 95% CI, 0.70-1.06; P = .19).
Health care–associated infections in all patients (with or without diabetes)
| Outcome measures | TXA (n = 307) | Placebo (n = 306) | RR (95% CI) | P |
|---|---|---|---|---|
| Primary end point | ||||
| Health care–associated infection | 79 (25.7) | 100 (32.7) | 0.79 (0.61-1.01) | .059 |
| Secondary end points* | 35 (11.4) | 36 (11.8) | 0.97 (0.63-1.50) | .89 |
| Pneumonia | ||||
| SSI | 44 (14.3) | 63 (20.6) | 0.70 (0.49-0.99) | .041 |
| Superficial | 37 (12.1) | 55 (18.0) | 0.67 (0.46-0.99) | .040 |
| Deep | 8 (2.6) | 9 (2.9) | 0.89 (0.35-2.27) | .80 |
| Organ space | 2 (0.7) | 3 (1.0) | 0.66 (0.11-3.95) | .69† |
| Sepsis | 37 (12.1) | 48 (15.7) | 0.87 (0.70-1.06) | 0.19 |
| Bacteremia | 2 (0.7) | 3 (1.0) | 0.66 (0.11-3.95) | 0.69† |
| Catheter line infection | 2 (0.7) | 1 (0.3) | 1.99 (0.18-21.9) | 0.99† |
| Outcome measures | TXA (n = 307) | Placebo (n = 306) | RR (95% CI) | P |
|---|---|---|---|---|
| Primary end point | ||||
| Health care–associated infection | 79 (25.7) | 100 (32.7) | 0.79 (0.61-1.01) | .059 |
| Secondary end points* | 35 (11.4) | 36 (11.8) | 0.97 (0.63-1.50) | .89 |
| Pneumonia | ||||
| SSI | 44 (14.3) | 63 (20.6) | 0.70 (0.49-0.99) | .041 |
| Superficial | 37 (12.1) | 55 (18.0) | 0.67 (0.46-0.99) | .040 |
| Deep | 8 (2.6) | 9 (2.9) | 0.89 (0.35-2.27) | .80 |
| Organ space | 2 (0.7) | 3 (1.0) | 0.66 (0.11-3.95) | .69† |
| Sepsis | 37 (12.1) | 48 (15.7) | 0.87 (0.70-1.06) | 0.19 |
| Bacteremia | 2 (0.7) | 3 (1.0) | 0.66 (0.11-3.95) | 0.69† |
| Catheter line infection | 2 (0.7) | 1 (0.3) | 1.99 (0.18-21.9) | 0.99† |
All data are n (%). Bold indicates significant results.
Patients could have >1 site of infection.
Fisher’s exact test.
The risk estimates for any infection with TXA were comparable in those who did (RR, 0.84) and did not (RR, 0.92) receive a blood transfusion within 24 hours of surgery (P for interaction = 0.54) and in those who did (RR, 0.86) and did not (RR, 0.82) receive a blood transfusion at any time during their hospitalization (P for interaction = 0.77). Similarly, the risk estimate for any infection with TXA was unaffected (RR, 0.79 vs 0.77) after adjusting for postoperative blood loss at 24 hour. Hence, the reduction in infection rates due to TXA cannot be explained by a difference in exposure to blood transfusion or blood loss.
Diabetes: a major confounding variable for infection risk
The scale of the ATACAS study allowed us to perform subgroup analyses to investigate the impact of diabetes on infection rates. Infection rates were nonsignificantly increased in placebo-treated diabetes patients vs nondiabetes patients (35% vs 32%; P = .55). However, TXA treatment profoundly reduced overall infection rates in nondiabetic patients compared with placebo (21% vs 32%; P = .017) (Table 2), but it failed to reduce infection rates in patients with diabetes compared with placebo (35% vs 35%; P = .99).
Health care–associated infections in patients without diabetes
| Outcome measures | TXA (n = 204) | Placebo (n = 200) | RR (95% CI) | P |
|---|---|---|---|---|
| Primary end point | ||||
| Health care–associated infection | 43 (21.1) | 63 (31.5) | 0.67 (0.48-0.94) | .017 |
| Secondary end points* | 23 (11.3) | 22 (11.0) | 1.03 (0.59-1.78) | .93 |
| Pneumonia | ||||
| SSI | 22 (10.8) | 36 (18.0) | 0.60 (0.37-0.98) | .039 |
| Superficial | 17 (8.3) | 32 (16.0) | 0.52 (0.30-0.91) | .018 |
| Deep | 4 (2.0) | 4 (2.0) | 0.98 (0.25-3.87) | .98 |
| Organ space | 1 (0.5) | 2 (1.0) | 0.49 (0.05-5.36) | .62† |
| Sepsis | 21 (10.3) | 31 (15.5) | 0.66 (0.40-1.12) | .12 |
| Bacteremia | 2 (1.0) | 1 (0.5) | 1.96 (0.18-21.5) | .57† |
| Catheter line infection | 2 (1.0) | 0 (0.0) | — | .50† |
| Outcome measures | TXA (n = 204) | Placebo (n = 200) | RR (95% CI) | P |
|---|---|---|---|---|
| Primary end point | ||||
| Health care–associated infection | 43 (21.1) | 63 (31.5) | 0.67 (0.48-0.94) | .017 |
| Secondary end points* | 23 (11.3) | 22 (11.0) | 1.03 (0.59-1.78) | .93 |
| Pneumonia | ||||
| SSI | 22 (10.8) | 36 (18.0) | 0.60 (0.37-0.98) | .039 |
| Superficial | 17 (8.3) | 32 (16.0) | 0.52 (0.30-0.91) | .018 |
| Deep | 4 (2.0) | 4 (2.0) | 0.98 (0.25-3.87) | .98 |
| Organ space | 1 (0.5) | 2 (1.0) | 0.49 (0.05-5.36) | .62† |
| Sepsis | 21 (10.3) | 31 (15.5) | 0.66 (0.40-1.12) | .12 |
| Bacteremia | 2 (1.0) | 1 (0.5) | 1.96 (0.18-21.5) | .57† |
| Catheter line infection | 2 (1.0) | 0 (0.0) | — | .50† |
All data are n (%). The dash indicates that the RR cannot be determined. Bold indicates significant results.
Patients could have >1 site of infection.
Fisher’s exact test.
We next evaluated the efficacy of TXA on transfusion needs in patients with or without diabetes in the entire ATACAS trial cohort12 (nondiabetes: 1512 TXA vs 1514 placebo; diabetes: 799 TXA vs 806 placebo) to maximize statistical power. Transfusion requirements were similar in the placebo groups of patients with or without diabetes (49% vs 49%; P = .89). TXA significantly reduced transfusion requirements within 24 hours postsurgery in the nondiabetes (P < .001) and diabetes (P < .001) cohorts. However, the effect of TXA was stronger in patients without diabetes (Table 3), with TXA reducing the RR for transfusion by 40% in nondiabetic patients but only by 28% in those with diabetes (P for interaction = .012). There also were significant reductions only in the patients without diabetes with regard to each of the secondary end points measuring blood loss, bleeding complications, and length of stay with TXA (all P < .05) (Table 3). Hence, diabetes promotes a partially refractory response to antifibrinolytic therapy.
Subgroup effects of TXA according to diabetes status in the ATACAS trial
| Patients without diabetes | Patients with diabetes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| TXA (n = 1512) | Placebo (n = 1514) | RR (95% CI) | P | TXA (n = 799) | Placebo (n = 806) | RR (95% CI) | P | Interaction P | |
| Outcome | |||||||||
| Total mediastinal drainage, median (IQR), mL | 820 (550-1250) | 1090 (750-1560) | — | <.001 | 730 (500-1130) | 1033 (700-1490) | — | <.001 | .41 |
| Blood transfusion up to 24 h after surgery, n (%) | 441 (29.2) | 739 (48.8) | 0.60 (0.54-0.66) | <.001 | 281 (35.2) | 391 (48.5) | 0.72 (0.64-0.82) | <.001 | .012 |
| Reoperation for bleeding or tamponade, n (%) | 22 (1.5) | 47 (3.1) | 0.47 (0.28-0.77) | .003 | 10 (1.3) | 18 (2.2) | 0.56 (0.26-1.21) | .14 | .70 |
| Total red cell transfusion, n (%), no. of units | 2 (1-3) | 2 (2-4) | — | <.001 | 2 (1-4) | 2 (2-4) | — | .074 | .31 |
| Length of stays* | HR (95% CI) | HR (95% CI) | |||||||
| ICU stay, h | 26 (21-54) | 29 (22-66) | 1.09 (1.01-1.17) | .022 | 40 (22-72) | 41 (22-71) | 1.01 (0.91-1.11) | .87 | .22 |
| Hospital stay, d | 8 (6-12) | 8 (6-14) | 1.09 (1.02-1.17) | .028 | 90 (7-14) | 9 (7-14) | 0.97 (0.88-1.07) | .54 | .074 |
| Patients without diabetes | Patients with diabetes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| TXA (n = 1512) | Placebo (n = 1514) | RR (95% CI) | P | TXA (n = 799) | Placebo (n = 806) | RR (95% CI) | P | Interaction P | |
| Outcome | |||||||||
| Total mediastinal drainage, median (IQR), mL | 820 (550-1250) | 1090 (750-1560) | — | <.001 | 730 (500-1130) | 1033 (700-1490) | — | <.001 | .41 |
| Blood transfusion up to 24 h after surgery, n (%) | 441 (29.2) | 739 (48.8) | 0.60 (0.54-0.66) | <.001 | 281 (35.2) | 391 (48.5) | 0.72 (0.64-0.82) | <.001 | .012 |
| Reoperation for bleeding or tamponade, n (%) | 22 (1.5) | 47 (3.1) | 0.47 (0.28-0.77) | .003 | 10 (1.3) | 18 (2.2) | 0.56 (0.26-1.21) | .14 | .70 |
| Total red cell transfusion, n (%), no. of units | 2 (1-3) | 2 (2-4) | — | <.001 | 2 (1-4) | 2 (2-4) | — | .074 | .31 |
| Length of stays* | HR (95% CI) | HR (95% CI) | |||||||
| ICU stay, h | 26 (21-54) | 29 (22-66) | 1.09 (1.01-1.17) | .022 | 40 (22-72) | 41 (22-71) | 1.01 (0.91-1.11) | .87 | .22 |
| Hospital stay, d | 8 (6-12) | 8 (6-14) | 1.09 (1.02-1.17) | .028 | 90 (7-14) | 9 (7-14) | 0.97 (0.88-1.07) | .54 | .074 |
These data have not been published previously. Bold indicates significant results.
HR, hazard ratio; ICU, intensive care unit; IQR, interquartile range.
Length-of-stay data are not normally distributed; therefore, these were analyzed as times to events using Cox regression. The hazard ratio is the risk for discharge on any given day: an HR > 1 indicates a higher rate of discharge, meaning a greater likelihood of discharge on a given day compared with placebo (ie, a shorter length of stay). The dash indicates that the risk ratio cannot be determined
Effect of cardiac surgery on innate immune cells and T cells
We next determined the temporal effect of TXA vs placebo administration on immune and inflammatory parameters in a subgroup of 41 consecutive cardiac surgery patients (n = 19 placebo; n = 22 TXA) recruited to the ATACAS trial.12
Cardiac surgery resulted in profound changes in the plasma cytokine and cellular immune characteristics in the placebo group. Eleven of the 14 myeloid and lymphoid cell subsets evaluated were significantly modulated as a consequence of surgery (supplemental Table 7). All 7 functional markers assessed on the myeloid subsets were affected by surgery (supplemental Table 8). Also, 4 of the 7 functional markers for lymphoid subpopulations were significantly altered by surgery (supplemental Table 9). Eight of the 10 cytokines evaluated were significantly changed by cardiac surgery (ie, IL-10 increased 130-fold, and IL-6 increased 50-fold, whereas the immunosuppressive cytokine TGF-β was reduced by ∼40%) (supplemental Table 10).
The effect of TXA on myeloid and lymphoid cell surface markers postsurgery
Despite the effect of cardiac surgery on the cellular immune profile, TXA treatment significantly modified a number of these immune cell parameters. Expression strength of the activation marker CD83 was increased relative to placebo-treated patients at POD-3 on classical, intermediate (supplemental Figure 2), and nonclassical monocytes, as well as on CD1c+ conventional dendritic cells (cDCs) (Figure 1A-B). Expression of the activation marker CCR7 on CD1c+ cDCs was downregulated in placebo-treated, but not in TXA-treated, patients at POD-3 (Figure 1C). CCR7 expression on plasmacytoid dendritic cells (pDCs) was selectively and significantly enhanced in TXA-treated patients at POD-1 (supplemental Figure 2). The programmed cell death–inducing marker PD-L1 was downregulated on nonclassical monocytes by TXA (Figure 1D). Expression of PD-L1 on CD1c+ cDCs was downregulated after surgery, with a trend toward a further reduction in expression strength in TXA-treated patients at 24 hours (supplemental Figure 2).

TXA modulates myeloid and lymphoid cells, as well as plasma cytokines, in the immune response after cardiac surgery. Patients undergoing cardiac surgery were treated with placebo or TXA, and the cellular immune response was characterized using flow cytometry before drug administration, as well as on POD-1 and POD-3. (A) CD83 expression was significantly increased on nonclassical monocytes in the TXA group at POD-3. (B) CD83 on CD1c+ cDCs was downregulated to a significantly lower extent in TXA-treated patients at POD-3. (C) TXA treatment also resulted in a significant relative increase in CCR7-expressing CD1c+ cDCs. (D) Moreover, TXA reduced expression of the programmed cell death inducing PD-L1 on nonclassical monocytes at POD-3. (E) CD4+ T cells were reduced significantly after cardiac surgery, whereby the decrease was significantly greater in the TXA group at POD-1. (F) CD8+ Tmem counts were also reduced at POD-1, but they remained reduced only in the TXA-treated patients at POD-3. Expression of the latent TGF-β releasing LAP (G) and CCR7 (H) relative to baseline was significantly lower in TXA-treated patients at POD-3. (I) In contrast, CCR7 expression on NK cells was significantly increased in the TXA group on POD-1. (J) Plasma levels of the proinflammatory cytokine IL-1β were significantly reduced in the TXA group compared with placebo at POD-1. Data represent fold-change from preoperative (preOP) levels and are expressed as mean ± standard error of the mean. Placebo: n = 19, TXA: n = 22. #P < .05, ##P < .01, ###P < .001, ####P < .0001 vs preOP, 1-way ANOVA with Dunnett’s correction test for multiple comparisons; *P < .05, placebo vs TXA, 2-tailed Student t test.
TXA modulates myeloid and lymphoid cells, as well as plasma cytokines, in the immune response after cardiac surgery. Patients undergoing cardiac surgery were treated with placebo or TXA, and the cellular immune response was characterized using flow cytometry before drug administration, as well as on POD-1 and POD-3. (A) CD83 expression was significantly increased on nonclassical monocytes in the TXA group at POD-3. (B) CD83 on CD1c+ cDCs was downregulated to a significantly lower extent in TXA-treated patients at POD-3. (C) TXA treatment also resulted in a significant relative increase in CCR7-expressing CD1c+ cDCs. (D) Moreover, TXA reduced expression of the programmed cell death inducing PD-L1 on nonclassical monocytes at POD-3. (E) CD4+ T cells were reduced significantly after cardiac surgery, whereby the decrease was significantly greater in the TXA group at POD-1. (F) CD8+ Tmem counts were also reduced at POD-1, but they remained reduced only in the TXA-treated patients at POD-3. Expression of the latent TGF-β releasing LAP (G) and CCR7 (H) relative to baseline was significantly lower in TXA-treated patients at POD-3. (I) In contrast, CCR7 expression on NK cells was significantly increased in the TXA group on POD-1. (J) Plasma levels of the proinflammatory cytokine IL-1β were significantly reduced in the TXA group compared with placebo at POD-1. Data represent fold-change from preoperative (preOP) levels and are expressed as mean ± standard error of the mean. Placebo: n = 19, TXA: n = 22. #P < .05, ##P < .01, ###P < .001, ####P < .0001 vs preOP, 1-way ANOVA with Dunnett’s correction test for multiple comparisons; *P < .05, placebo vs TXA, 2-tailed Student t test.
TXA significantly reduced the levels of CD4+ T cells, CD8+ memory T cells (Tmems) (Figure 1E-F), and it attenuated expression of the suppressive (TGF-β–releasing) molecule LAP, as well as CCR7, on regulatory T cells (Tregs) (Figure 1G-H). Conversely, TXA administration increased CCR7 expression on natural killer (NK) cells (Figure 1I).
Together, these data show that TXA upregulates immune-enhancing markers and downregulates immunosuppressive markers in cardiac surgery patients, suggestive of improved immune competence. It is also important to note that this subgroup well reflected our observations seen in the entire study population of 613 patients, because TXA treatment significantly reduced infection rates in nondiabetic patients (P = .03).
TXA selectively induces changes to the cellular immune profile in nondiabetic cardiac surgery patients
We next compared the effect of TXA on immune marker levels in nondiabetic and diabetic patients. Of the 41 patients evaluated in Figure 1, 14 were diabetic (8 placebo, 6 TXA). In nondiabetic patients, the activation marker CD83 was enhanced by TXA on several myeloid subsets (classical [Figure 2A] and intermediate [Figure 2B] monocytes and CD1c+ cDCs [Figure 2C]) at POD-3, whereas the immunosuppressive marker PD-L1 was downregulated on CD1c+ cDCs (Figure 2D) and nonclassical monocytes (Figure 2E), consistent with our findings on infection outcome. In contrast, these effects were not present in the diabetic cohort (Figure 2G-L). Indeed, PD-L1 even shows increased expression (significant in nonclassical monocytes [Figure 2K] and not significant in CD1c+ cDCs [Figure 2J]) at POD-1. Furthermore, expression of the immunosuppressive marker LAP on Tregs was significantly reduced by TXA on POD-3 in nondiabetic (Figure 2F), but not in diabetic (Figure 2L), patients. Hence, patients with diabetes are refractory to the stimulatory effects of TXA on the innate cellular immune system.
![Figure 2. TXA induces changes to the cellular immune profile in cardiac surgery patients, consistent with enhanced immune activation, selectively in patients without diabetes. Results obtained with flow cytometry were analyzed separately for diabetic and nondiabetic patients. Consistent with our findings on infection outcome, the activation marker CD83 was enhanced by TXA on several myeloid subsets (classical [A] and intermediate [B] monocytes and CD1c+ cDCs [C]) at POD-3 in patients without diabetes, whereas the immunosuppressive marker PD-L1 was downregulated on CD1c+ cDCs (D) and nonclassical monocytes (E). (G-L) In contrast, these effects were not seen in the diabetic cohort. In fact, PD-L1 even exhibited increased expression (significant in nonclassical monocytes [K], nonsignificant in CD1c+ cDCs [J]) at POD-1. In addition, expression of the immunosuppressive marker LAP on Tregs was significantly reduced by TXA on POD-3 in patients without diabetes (F) but not in patients with diabetes (L). Data represent fold-change from preoperative (preOP) levels and are expressed as mean ± standard error of the mean. Placebo: n = 11, TXA: n = 16 (patients without diabetes); placebo: n = 8, TXA: n = 6 (patients with diabetes). *P < .05, **P < .01, 2-tailed Student t test. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/10/10.1182_bloodadvances.2019000092/2/m_advancesadv2019000092f2.png?Expires=1765883727&Signature=vB-~8wV1WNId0bbR614tfy-XuzrzcUL5gkdnJYJ2KizMvKBOeLbHE5YedndjPewIlFNLyrp2Nmu1ymq3okUD4VKbNi8vARXsPG70axcaHJWnH6OrMosigbXTiD~KgxoWbTZmOBTusN3oNLDptI5OX59qz3s4MH2EuK06qtaqG4FY83FV1yZsRPT3vuQMZ6oTcgEFrUXnnDr3DOnuht1b2FNZofKx~93GqCVHOWuJq5xsayTavQdcY9lGbXQly~Q52b2ZBiWT-8kLrSXzocgWV3m4o9gnZxPL3vChDNxtm8gH32X0DB822nAApZlG3RYv3cDB7IOQpmBx1rrUaRkmOw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TXA induces changes to the cellular immune profile in cardiac surgery patients, consistent with enhanced immune activation, selectively in patients without diabetes. Results obtained with flow cytometry were analyzed separately for diabetic and nondiabetic patients. Consistent with our findings on infection outcome, the activation marker CD83 was enhanced by TXA on several myeloid subsets (classical [A] and intermediate [B] monocytes and CD1c+ cDCs [C]) at POD-3 in patients without diabetes, whereas the immunosuppressive marker PD-L1 was downregulated on CD1c+ cDCs (D) and nonclassical monocytes (E). (G-L) In contrast, these effects were not seen in the diabetic cohort. In fact, PD-L1 even exhibited increased expression (significant in nonclassical monocytes [K], nonsignificant in CD1c+ cDCs [J]) at POD-1. In addition, expression of the immunosuppressive marker LAP on Tregs was significantly reduced by TXA on POD-3 in patients without diabetes (F) but not in patients with diabetes (L). Data represent fold-change from preoperative (preOP) levels and are expressed as mean ± standard error of the mean. Placebo: n = 11, TXA: n = 16 (patients without diabetes); placebo: n = 8, TXA: n = 6 (patients with diabetes). *P < .05, **P < .01, 2-tailed Student t test. ns, not significant.
TXA induces changes to the cellular immune profile in cardiac surgery patients, consistent with enhanced immune activation, selectively in patients without diabetes. Results obtained with flow cytometry were analyzed separately for diabetic and nondiabetic patients. Consistent with our findings on infection outcome, the activation marker CD83 was enhanced by TXA on several myeloid subsets (classical [A] and intermediate [B] monocytes and CD1c+ cDCs [C]) at POD-3 in patients without diabetes, whereas the immunosuppressive marker PD-L1 was downregulated on CD1c+ cDCs (D) and nonclassical monocytes (E). (G-L) In contrast, these effects were not seen in the diabetic cohort. In fact, PD-L1 even exhibited increased expression (significant in nonclassical monocytes [K], nonsignificant in CD1c+ cDCs [J]) at POD-1. In addition, expression of the immunosuppressive marker LAP on Tregs was significantly reduced by TXA on POD-3 in patients without diabetes (F) but not in patients with diabetes (L). Data represent fold-change from preoperative (preOP) levels and are expressed as mean ± standard error of the mean. Placebo: n = 11, TXA: n = 16 (patients without diabetes); placebo: n = 8, TXA: n = 6 (patients with diabetes). *P < .05, **P < .01, 2-tailed Student t test. ns, not significant.
Effect of TXA on the cytokine response postsurgery
TXA treatment attenuated the surgery-induced increase in levels of the proinflammatory cytokine IL-1β at POD-1 compared with placebo (Figure 1J), but it did not significantly alter levels of TNF-α, IL-6, IL-8, MCP-1, IFN-γ, IL-13, IL-10, TGF-β, or sTNFR2 (data not shown).
TXA blocks plasmin activity in cardiac surgery
Cardiac surgery is known to increase levels of D-dimer as a result of plasmin-mediated fibrinolysis.14,15 Accordingly, D-dimer levels were significantly increased postoperatively and remained elevated up to POD-3 in the placebo group. However, TXA completely inhibited the postoperative increase in D-dimer, which was only significantly elevated on POD-3, most likely because TXA had been cleared at this point. Hence, plasmin activity postsurgery was effectively inhibited by TXA (supplemental Figure 3).
Effect of TXA on the cellular immune profile in healthy volunteers
We next determined the temporal effect of 1 g of TXA on levels of plasma cytokines and the cellular immune profile in healthy volunteers. TXA significantly reduced plasma levels of proinflammatory cytokines, with a decrease in TNF-α at 4 hours, as well as in IL-6 between 2 and 24 hours (Figure 3). IFN-γ levels were reduced significantly at 2 hours and 4 hours, whereas the levels of IL-10 were reduced significantly only at 2 hours (Figure 3Ai). sTNFR2 levels were significantly enhanced by TXA at 2 and 4 hours but declined again at 24 hours (Figure 3Aii). Other cytokines (IL-1β, IL-8, MCP-1, IL-13, TGF-β) were not changed (supplemental Table 11).

TXA changes plasma cytokine levels and functional marker expression on myeloid cell populations in healthy volunteers. Plasma cytokine levels and expression of functional markers on myeloid cells were evaluated in healthy volunteers at various time points after administration of TXA. TXA significantly reduced plasma levels of the proinflammatory cytokine TNF-α at 4 hours, as well as IL-6 between 2 hours and 24 hours. (Ai) Levels of the type 1 T helper cell cytokine IFN-γ were significantly reduced at 2 hours and 4 hours, and the levels of the type 2 T helper cell cytokine IL-10 were significantly reduced at 2 hours (n = 9). (Aii) sTNFR2 levels were significantly enhanced by TXA at 2 hours and 4 hours but returned to baseline at 24 hours (n = 9). (B) Levels of circulating nonclassical monocytes were significantly enhanced by TXA after 24 hours, whereas pDC levels were reduced after 4 hours, yet returned to baseline levels after 24 hours (n = 6). (C) TXA significantly increased expression of the activation marker CD83 on classical and intermediate monocytes, as well as on CD1c+ cDCs (n = 6). (D) The maturation marker HLA-DR was reduced at 24 hours on intermediate monocytes (already at 4 hours), classical monocytes, and pDCs (n = 6). (E) CCR7, indicating migration to secondary lymphatic organs, was significantly reduced by TXA in classical and intermediate monocytes (n = 6). (F) TNFR2, mediating TNF-α signaling, was increased at 4 hours and 24 hours in classical and intermediate monocytes and CD1c+ cDCs, as well as in pDCs at 24 hours after TXA intake (n = 6). (G) PD-L1, which induces programmed cell death in effector cells of the immune system, was significantly downregulated by TXA after 4 hours and even further after 24 hours in classical monocytes and intermediate monocytes, as well as in CD1c+ cDCs and the immunosuppressive monocytic myeloid-derived suppressor cell (MO-MDSC) at 24 hours (n = 6). Data are expressed as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, repeated measures 1-way ANOVA with Dunnett’s correction test for multiple comparisons.
TXA changes plasma cytokine levels and functional marker expression on myeloid cell populations in healthy volunteers. Plasma cytokine levels and expression of functional markers on myeloid cells were evaluated in healthy volunteers at various time points after administration of TXA. TXA significantly reduced plasma levels of the proinflammatory cytokine TNF-α at 4 hours, as well as IL-6 between 2 hours and 24 hours. (Ai) Levels of the type 1 T helper cell cytokine IFN-γ were significantly reduced at 2 hours and 4 hours, and the levels of the type 2 T helper cell cytokine IL-10 were significantly reduced at 2 hours (n = 9). (Aii) sTNFR2 levels were significantly enhanced by TXA at 2 hours and 4 hours but returned to baseline at 24 hours (n = 9). (B) Levels of circulating nonclassical monocytes were significantly enhanced by TXA after 24 hours, whereas pDC levels were reduced after 4 hours, yet returned to baseline levels after 24 hours (n = 6). (C) TXA significantly increased expression of the activation marker CD83 on classical and intermediate monocytes, as well as on CD1c+ cDCs (n = 6). (D) The maturation marker HLA-DR was reduced at 24 hours on intermediate monocytes (already at 4 hours), classical monocytes, and pDCs (n = 6). (E) CCR7, indicating migration to secondary lymphatic organs, was significantly reduced by TXA in classical and intermediate monocytes (n = 6). (F) TNFR2, mediating TNF-α signaling, was increased at 4 hours and 24 hours in classical and intermediate monocytes and CD1c+ cDCs, as well as in pDCs at 24 hours after TXA intake (n = 6). (G) PD-L1, which induces programmed cell death in effector cells of the immune system, was significantly downregulated by TXA after 4 hours and even further after 24 hours in classical monocytes and intermediate monocytes, as well as in CD1c+ cDCs and the immunosuppressive monocytic myeloid-derived suppressor cell (MO-MDSC) at 24 hours (n = 6). Data are expressed as mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, repeated measures 1-way ANOVA with Dunnett’s correction test for multiple comparisons.
Blood was also evaluated for changes in multiple innate immune cells and T cell subsets at 4 and 24 hours after TXA administration. TXA did not result in significant changes in the numbers of the evaluated cell subsets at the 4-hour time point, with the exception of pDCs, which were transiently reduced by >50% (Figure 3B). At the 24-hour time point, there were significant increases in the levels of nonclassical monocytes (Figure 3B) and T cells overall, with selective increases in CD4+ and CD8+ T cells, as well as immunosuppressive Tregs (Figure 4A). Classical monocytes and CD8+ effector T cells (Teffs) were also increased at 24 hours, albeit not significantly (supplemental Table 12).
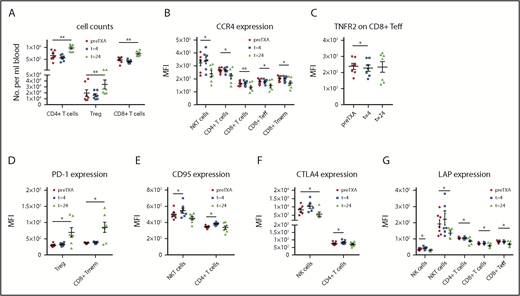
TXA changes levels of T cell subsets and functional marker expression on lymphoid cell populations in healthy volunteers. Expression of functional markers on lymphoid subsets was evaluated in healthy volunteers at various time points after administration of TXA. (A) A marked increase was observed in the levels of CD4+ T cells, including Tregs and CD8+ T cells (n = 7). (B) CCR4, which mediates migration to sites of inflammation, was upregulated after 24 hours in NKT cells, CD4+ T cells, CD8+ T cells, and the CD8+ Teff and Tmem subsets (n = 7). (C) TNFR2 expression was significantly reduced on CD8+ Teffs after 4 hours, yet it returned to normal levels after 24 hours (n = 7). (D) Expression of PD-1, the receptor of PD-L1, which mediates programmed cell death upon activation, was enhanced on Tregs and CD8+ Tmems (n = 7). (E) CD95, another death receptor, was rapidly upregulated by TXA on NKT cells and CD4+ T cells after 4 hours but had returned to normal levels at the 24-hour time point (n = 7). (F) Augmented CTLA4 expression was evident on CD4+ T cells at 4 hours and on NK cells at 24 hours post-TXA intake (n = 7). (G) The TGF-β releasing LAP was expressed significantly more weakly on NK and NKT cells, as well as CD4+ and CD8+ T cells, including CD8+ Teffs, 24 hours after TXA intake (n = 7). Data are expressed as mean ± standard error of the mean. *P < .05, **P < .01, repeated-measures 1-way ANOVA with Dunnett’s correction test for multiple comparisons.
TXA changes levels of T cell subsets and functional marker expression on lymphoid cell populations in healthy volunteers. Expression of functional markers on lymphoid subsets was evaluated in healthy volunteers at various time points after administration of TXA. (A) A marked increase was observed in the levels of CD4+ T cells, including Tregs and CD8+ T cells (n = 7). (B) CCR4, which mediates migration to sites of inflammation, was upregulated after 24 hours in NKT cells, CD4+ T cells, CD8+ T cells, and the CD8+ Teff and Tmem subsets (n = 7). (C) TNFR2 expression was significantly reduced on CD8+ Teffs after 4 hours, yet it returned to normal levels after 24 hours (n = 7). (D) Expression of PD-1, the receptor of PD-L1, which mediates programmed cell death upon activation, was enhanced on Tregs and CD8+ Tmems (n = 7). (E) CD95, another death receptor, was rapidly upregulated by TXA on NKT cells and CD4+ T cells after 4 hours but had returned to normal levels at the 24-hour time point (n = 7). (F) Augmented CTLA4 expression was evident on CD4+ T cells at 4 hours and on NK cells at 24 hours post-TXA intake (n = 7). (G) The TGF-β releasing LAP was expressed significantly more weakly on NK and NKT cells, as well as CD4+ and CD8+ T cells, including CD8+ Teffs, 24 hours after TXA intake (n = 7). Data are expressed as mean ± standard error of the mean. *P < .05, **P < .01, repeated-measures 1-way ANOVA with Dunnett’s correction test for multiple comparisons.
TXA significantly increased expression of the activation marker CD83 on classical and intermediate monocytes, as well as on CD1c+ cDCs (Figure 3C). CD86 was not altered in any of the assessed cell subsets (supplemental Table 13). In contrast to CD83, surface expression of the maturation marker HLA-DR was reduced after 24 hours on intermediate monocytes (already at 4 hours) classical monocytes, and pDCs (Figure 3D). The inflammatory site–associated migration marker CCR4 was unchanged in all assessed myeloid subsets. In contrast, CCR7, which promotes migration to secondary lymphatic organs, was significantly reduced by TXA on classical and intermediate monocytes (Figure 3E). TNFR2, mediating TNF-α signaling, was increased at 4 and 24 hours in classical and intermediate monocytes, as well as on CD1c+ cDCs and pDCs at 24 hours after TXA intake (Figure 3F). PD-L1 was significantly downregulated by TXA after 4 hours and even further after 24 hours on classical monocytes and intermediate monocytes, as well as on CD1c+ cDCs and on the immunosuppressive MO-MDSC subset at 24 hours (Figure 3G).
Many lymphoid markers associated with immunosuppression, migration, and cytokine signaling were significantly altered by TXA (Figure 4). For example, CCR4, which mediates migration to sites of inflammation, was downregulated after 24 hours in natural killer T (NKT) cells, CD4+ T cells, CD8+ T cells, and the CD8+ Teff and Tmem subsets (Figure 4B). In contrast, CCR7 was not changed in any of the assessed lymphocyte subsets (supplemental Table 14). TNFR2 expression was mildly, but significantly, reduced on CD8+ Teffs after 4 hours, yet it returned to normal levels after 24 hours (Figure 4C). Expression of PD-1, the receptor of PD-L1, was enhanced on Tregs and CD8+ Tmems (Figure 4D). Upregulation was also observed in NKT cells, although this was not significant (supplemental Table 14). CD95, also a death receptor, was rapidly upregulated by TXA on NKT cells and CD4+ T cells after 4 hours but returned to normal levels again at the 24-hour time point (Figure 4E). CTLA4 expression was significantly reduced on NK cells at 24 hours post-TXA intake. Augmented CTLA4 expression was also evident on CD4+ T cells at 4 hours (Figure 4F). LAP expression was significantly reduced by TXA on NK cells, NKT cells, CD4+ T cells, and CD8+ T cells, including CD8+ Teffs, after 24 hours (Figure 4G).
Hence, administration of the normal clinical dosage of TXA in healthy volunteers causes significant changes in multiple innate immune cells and T cell subsets, in addition to reducing proinflammatory cytokine levels, while increasing sTNFR2, which binds and inhibits TNF-α.16
A summary of the effects of TXA on the immune response is provided in Figure 5.
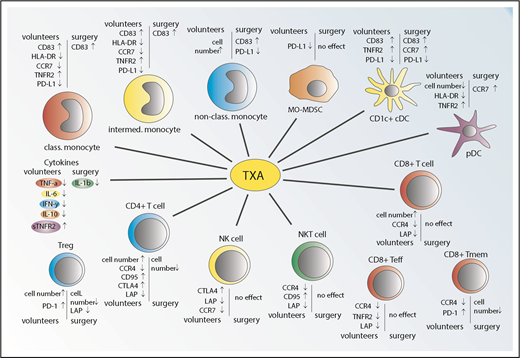
Summary of the effects of TXA treatment in healthy volunteers and surgery patients. TXA administration causes marked alteration in a variety of plasma cytokine levels, numbers of immune cells, and/or expression of key markers associated with immune activation and immunosuppression.
Summary of the effects of TXA treatment in healthy volunteers and surgery patients. TXA administration causes marked alteration in a variety of plasma cytokine levels, numbers of immune cells, and/or expression of key markers associated with immune activation and immunosuppression.
Discussion
The immunosuppressive effect of plasmin in animal models and on human cells in vitro8 led us to explore the consequences of plasmin blockade by TXA on the immune response in patients undergoing cardiac surgery and in healthy volunteers. Although it was not our expectation that TXA would produce identical changes in the immune cell profile in volunteers and cardiac surgery patients, given that the surgical procedure alone would have induced a profound effect, it is very interesting to note that TXA did indeed increase the activation marker CD83, while simultaneously decreasing the key immunosuppressive marker PD-L1, on multiple myeloid cell subsets in patients and volunteers. These changes are consistent with TXA administration causing a more robust immune response. Supporting this key finding, TXA resulted in a significant reduction in the frequency of postsurgery infection rates, despite administration of prophylactic antibiotic treatment.
Because the current indication for TXA administration is to reduce bleeding and the need for transfusion, we considered the possibility that the reduction in postsurgical infection rates by TXA was due to its hemostatic effects.17 However, the beneficial effect of TXA was independent of transfusion, as well as its effect on blood loss. Therefore, we propose that the reduction in infection rates by TXA was due to its capacity to counteract the early immunosuppressive action of plasmin, formed as a consequence of surgical trauma. Our evaluation of D-dimer levels confirmed that plasmin activity had been significantly increased postsurgery, but this did not occur in patients administered TXA.
The confounding effect of diabetes
We evaluated the effects of TXA on infection rates and transfusion requirements/blood loss in patients with and without diabetes because of the association of diabetes with increased infection risk and the fact that the fibrinolytic system is impaired in diabetes. This occurs not only because of increased levels of antifibrinolytic proteins18 but also because plasminogen is glycated and poorly activatable.19 This line of investigation led to 2 critically important findings. The first was the complete failure of TXA to reduce postoperative infection rates in patients with diabetes; indeed, this analysis more impressively highlighted the ability of TXA to reduce infection rates in nondiabetic patients in whom health care–associated infection rates overall were reduced by >30%. Importantly, TXA failed to modulate expression of the classic immune markers CD83, PD-L1, and LAP in the diabetic cohort. Although TXA significantly reduced blood transfusion needs in patients with diabetes, this effect was significantly less than the one seen in nondiabetic patients. Hence, this weaker effect of TXA in patients with diabetes may explain why TXA was ineffective at reducing infection rates in this patient cohort. On the other hand, the effect of TXA on mediastinal drainage was similar in the diabetic and nondiabetic groups, suggesting that TXA was stabilizing local fibrin deposits similarly in the 2 groups. This supports our hypothesis that the differential effect of TXA on infection rates in patients with diabetes is not related to the hemostatic effect of this drug.
This is the first study to report that diabetic patients are partially refractory to TXA treatment, suggesting that other antifibrinolytic approaches might need to be considered for patients with diabetes prior to surgery.
Immune modulation in healthy volunteers
The final aspect of our study was to evaluate the effect of TXA in healthy volunteers to avoid the confounding effects of bleeding and surgical trauma. Volunteers were administered the more common dosage (1 g) of TXA (ie, as used for tooth extractions in hemophilia patients).20 Despite the lower dosage compared with the cardiac surgery cohort, TXA still significantly modulated various cytokines (ie, TNF-α, IL-6, IFN-γ, IL-10) and cellular immune markers (ie, CD83, HLA-DR, CCR4, CCR7, TNFR2, PD-L1, PD-1, CD95, CTLA-4, LAP), further indicating a role for steady-state plasmin levels in the promotion of a quiescent immune state.13 Given the significant increase in circulating CD4+ and CD8+ T cells at 24 hours postadministration, the counteracting effect of TXA is also consistent with an immune-priming effect, creating the potential for a more responsive immune system. These changes were specific to TXA and not due to circadian fluctuations, because results were consistent across the participants in this study, and volunteers not treated with TXA did not display any differences over 24 hours in our previously published validation experiments.13
An important question relates to how TXA is implementing these effects on the immune response. Plasminogen is known to be essential for wound repair.21 Moreover, plasminogen supplementation has been shown to accelerate wound-repair processes.22-24 Hence, inhibition of plasminogen activation by TXA, albeit transiently, may have temporarily retarded the wound-repair process, potentially increasing infection risk; however, because the opposite effect was seen, this is unlikely.
The potential immunological effects of TXA are diverse, but lysine binding site–dependent plasmin generation on the cell surface has been shown to be inhibited by TXA.25,26 More than 12 plasminogen receptors have been described,27 with many expressed on monocytes and macrophages, including the recently described Plg-RKT.28 Plasminogen binding to this receptor (and various other receptors) is lysine dependent and blocked using lysine analogs.29 Plg-RKT is also a transmembrane protein containing a cytoplasmic tail that can potentially initiate intracellular signaling and influence cellular responses when bound to plasminogen28 ; this would also be blocked by TXA. Our D-dimer data show that the concentration of TXA used in our study inhibited fibrinolysis; hence, lysine-dependent binding of plasminogen to fibrin was also blocked. Therefore, it is likely that binding of plasminogen to ≥1 of its receptors via a lysine binding site, which, in turn, would have immunosuppressive consequences, would also have been inhibited. It would be interesting to determine whether cell surface plasminogen binding was reduced by TXA to support this notion.
Another key question is whether the TXA-dependent modulation of the markers of the innate immune response is actually causative in relation to infection risk. Although highly plausible, this still has not been demonstrated directly. Our finding that TXA modulated expression levels of the same markers in healthy volunteers not undergoing surgical trauma or experiencing infection further strengthens the hypothesis that these immune changes precede the onset of infection.
In summary, TXA administration changed plasma cytokine levels, as well as the number and surface marker expression of circulating leukocytes. Among at-risk patients undergoing coronary artery surgery, the use of TXA significantly reduced postoperative infection rates in patients without diabetes, independent of its effect on transfusion and blood loss. This finding has profound clinical implications that could potentially broaden the scope of TXA usage.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Catherine Farrington, Wendy Gallagher, and Andrea Ditoro (Department of Anaesthesia and Perioperative Medicine, Alfred Hospital) for support; Peter Grant (Faculty of Medicine and Health, University of Leeds) for insightful discussions; the Alfred Medical Research and Education Precinct (AMREP) Flow Cytometry Core Facility for technical assistance with the flow cytometry experiments; the volunteers for their participation and blood donation for this investigation; and Tom Chung (Crux Biolab) for performing the multiplex cytokine assays.
This work was supported by grant APP1045755 from the National Health and Medical Research Council (NHMRC) of Australia (R.L.M.) and by the Alfred Foundation. The healthy volunteer study was funded by the Australian Haemophilia Centre Directors Organisation. P.S.M. is an NHMRC Practitioner Fellow, M.P. is an NHMRC Senior Research Fellow, and R.L.M. is an NHMRC Principal Research Fellow.
Authorship
Contribution: D.F.D. conceived the project, performed experiments, analyzed data, collected and processed blood, and wrote and edited the manuscript; K.Y. and A.W. performed clinical data collection and analysis; S.W. and L.M.W. performed clinical sample collection and provided logistic support for the trial; G.H. collected and processed blood and processed data; M.D. and H.H. performed experiments and processed data; A.F. performed statistical analysis; M.P. designed flow cytometry experiments, analyzed data, and edited the manuscript; I.G. collected blood samples and provided logistics support for volunteer blood sample collection; H.T. collected blood samples, provided logistics support for volunteer blood sample collection, and edited the manuscript; M.S. conceived the project; P.S.M. conceived the project, analyzed data, provided logistics support for the trial, performed statistical analysis, and wrote and edited the manuscript; and R.L.M. conceived the project, analyzed data, and wrote and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert L. Medcalf, Molecular Neurotrauma and Haemostasis, Australian Centre for Blood Diseases, Monash University, Melbourne, VIC 3004, Australia; e-mail: robert.medcalf@monash.edu.

