

Key Points
Fluorescent labeling of CRISPR/Cas9–gRNA RNP enables sorting of edited HSPCs and iPSCs for further applications.
GADD45B plays a crucial role in UV stress-induced response of HSPCs and iPSCs.

Abstract
CRISPR/Cas9-mediated gene editing of stem cells and primary cell types has several limitations for clinical applications. The direct delivery of ribonucleoprotein (RNP) complexes consisting of Cas9 nuclease and guide RNA (gRNA) has improved DNA- and virus-free gene modifications, but it does not enable the essential enrichment of the gene-edited cells. Here, we established a protocol for the fluorescent labeling and delivery of CRISPR/Cas9–gRNA RNP in primary human hematopoietic stem and progenitor cells (HSPCs) and induced pluripotent stem cells (iPSCs). As a proof of principle for genes with low-abundance transcripts and context-dependent inducible expression, we successfully deleted growth arrest and DNA-damage-inducible β (GADD45B). We found that GADD45B is indispensable for DNA damage protection and survival in stem cells. Thus, we describe an easy and efficient protocol of DNA-free gene editing of hard-to-target transcripts and enrichment of gene-modified cells that are generally difficult to transfect.
Introduction
CRISPR/Cas9-mediated gene editing1,2 has a tremendous potential for clinical applications, such as gene therapy of inherited disorders or boosting of immune cells for cancer immunotherapies.3-9 Several monogenic disorders, including life-threatening bone marrow failure syndromes, might be treated by CRISPR/Cas9-mediated gene correction in autologous hematopoietic stem and progenitor cells (HSPCs) ex vivo. This procedure could then be followed by transplantation of the corrected HSPCs without exposing the patient to harsh immunosuppression regimens (ie, chemotherapy, irradiation).
Two groups recently published successful gene therapy approaches to cure sickle cell disease, a common inherited blood disorder. They generated deletions in the β-globin gene locus using CRISPR/Cas9 technology to mimic the hereditary persistence of fetal hemoglobin mutations.10,11 First attempts at gene editing using CRISPR/Cas9 for cancer therapy have also been launched recently. CRISPR/Cas9-generated chimeric antigen receptor–modified T cells targeting the checkpoint receptor programmed cell death 1 have been injected in a patient with metastatic non-small cell lung cancer.12,13 In addition to HSPCs, CRISPR/Cas9-mediated gene editing was successfully applied in neurons, hepatocytes, and cardiomyocytes.14-20
To further advance clinical applications of the CRISPR/Cas9 technology, unspecific integrations of viral or plasmid CRISPR/Cas9 DNA in the host genome and undesirable immune responses must be prevented. This may be achieved by transient virus- and DNA-free delivery approaches using CRISPR/Cas9–guide RNA (gRNA) ribonucleoprotein (RNP) complexes. Compared with DNA-based approaches, the direct delivery of CRISPR/Cas9–gRNA RNP complexes might have several advantages. Due to the cellular degradation of the RNP complex, exposure of cells to Cas9 is generally only transient and restricted, thereby limiting potential off-target effects of endonuclease overexpression.
Gundry et al recently described the efficient delivery of CRISPR/Cas9–gRNA RNP in HSPCs21 ; however, the method used in that study did not allow the enrichment of gene-edited cells. Purification of gene-edited HSPCs early in the manufacturing process is desirable, especially for clinical applications, because HSPCs differentiate and progressively lose their long-term repopulating capacity during culture. This is especially true for genes with low-abundance messenger RNA (mRNA) transcripts or inducible mRNA expression. These genes may be difficult to target, also due to epigenetic modifications, leading to tightly packed chromatin at the time of gRNA delivery. In these cases, the selection of gene-edited cells is indispensable. Nonmodified cells may retain a proliferative advantage over gene-edited cells, especially in mixed populations.
Introduction of a fluorescent tag to the CRISPR/Cas9-gRNA RNP enables enrichment of gene-edited cells for further experimental and clinical applications. Tagging of Cas9 protein could be achieved by fusion of Cas9 with fluorescent proteins or by chemical labeling of the CRISPR/Cas9–gRNA RNP with a fluorescent dye. However, Cas9–GFP fusion proteins might affect the intracellular localization, activity, or on- and off-target specificity of the endonuclease. Therefore, in the present study, we established a safe, simple, and efficient method for CRISPR/Cas9 gene knockout using transfection of stem cells with fluorescently labeled CRISPR/Cas9–gRNA RNP complexes.
Materials and methods
Cell culture
Human embryonic kidney 293FT (HEK293FT) and Jurkat cells were cultured under standard conditions (37°C, 5% CO2) using Dulbecco’s modified Eagle medium high glucose (HEK293FT cells) or RPMI 1640 GlutaMAX (Jurkat cells) medium supplemented with 10% fetal calf serum (Sigma-Aldrich) and 1% penicillin/streptomycin (Biochrome). HEK293FT cells were detached using 0.05% Trypsin-EDTA (Gibco) and seeded at a density of 1 × 105 cells per milliliter of medium. Jurkat cells were seeded at a density of 1 to 2 × 105 cells per milliliter of medium.
Human CD34+ HSPCs were isolated from the bone marrow or leukapheresis mononuclear cell fraction by magnetic bead separation (Human CD34 Progenitor Cell Isolation kit; Miltenyi Biotech). CD34+ cells were cultured in a density of 2 × 105 cells/mL of Stemline II medium (Sigma Aldrich) supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, 1% L-glutamine, and a cytokine cocktail consisting of 20 ng/mL IL-3, 20 ng/mL IL-6, 20 ng/mL thrombopoietin, 50 ng/mL SCF, and 50 ng/mL Flt-3L. Human induced pluripotent stem cells (iPSCs) were cultured on plates coated with Geltrex lactate dehydrogenase elevating virus–free reduced growth factor basement membrane matrix (cat. no. A1413201; Thermo Fisher Scientific) at a density of 2 × 105 cells/mL in StemFlex medium (cat. no. A3349401; Thermo Fisher Scientific) supplemented with 1% penicillin/streptomycin.
Generation and testing of the GADD45B gRNA
Specific CRISPR RNA (crRNA) for the first exon of the GADD45B gene (GCTCGTGGCGTGCGACAACGCGG, cut site: chr19 [+2,476,389: −2,476,389], NM_015675.3 Exon 1, 31bp; NP_056490.2 position N11) was designed using an online tool from the University of Heidelberg (http://crispr.cos.uni-heidelberg.de). The crRNA for GADD45B was first tested in transfected HEK293FT cells showing a gene modification efficiency of 67% in the total population of transfected cells.
Labeling of gRNA and plasmid DNA
Trans-activating CRISPR RNA (tracrRNA) and crRNA, obtained from IDT, were annealed at a ratio of 1:1 by incubating for 15 minutes at room temperature to generate gRNA. gRNA was fluorescently labeled using LabelIT CX-Rhodamine (cat. no. MIR7022; Mirus) or LabelIT Fluorescein (cat. no. MIR7025; Mirus) kits according to the manufacturer’s instructions. Labeling reagent and nucleic acid ratio were used at a ratio of 1:1 leading to 1 label per 20 to 60 bases, which is suitable for most applications.
Generation of crRNA-tracrRNA duplexes (gRNA) was conducted by adding 800 pmol of GADD45B-targeting crRNA and 800 pmol tracrRNA into 40 µL nuclease-free duplex buffer (IDT) at room temperature for 15 minutes. Labeling of the gRNA was performed by mixing gRNA, DNase- and RNase-free water, 10× labeling buffer A and 1:10 of LabelIT Reagent (CX-rhodamine or fluorescein). The reaction was incubated at 37°C for 1 hour while centrifuging briefly after 30 minutes to minimize evaporation and maintain the appropriate concentration of the reaction components.
Purification of labeled gRNA was conducted using the ethanol precipitation method. To this end, 5 M sodium chloride (0.1 volume) and ice-cold ethanol (2.5 volume) were added to the reaction, mixed well, and placed at −20°C for 30 minutes. Afterwards, the sample was centrifuged at 14 000g at 4°C for 30 minutes to pellet the labeled gRNA. Once pelleted, the supernatant was discarded gently without disturbing the pellet. The pellet was washed using 70% ethanol at room temperature and centrifuged at 14 000g for 30 minutes. After centrifugation, the pellet was air dried for 5 minutes and resolved in IDT nuclease-free duplex buffer. The labeled gRNA stock was stored at −20°C for up to 2 months.
Labeling of the pMAX GFP plasmid (Lonza) was carried out using LabelIT Tracker Intracellular Nucleic Acid Localization Kit (cat. no. MIR7022; Mirus) following the manufacturer’s protocol.
Assessment of the RNA integrity using Agilent Bioanalyzer
Labeled and unlabeled gRNA were analyzed using the Agilent RNA 6000 Pico Kit according to the manufacturer's instructions on the Agilent 2100 Bioanalyzer using the total RNA program.
Transfection of cells with CRISPR/Cas9-gRNA RNP complexes
Transfection was carried out either using TransIT-X2 (cat. no. MIR6003; Mirus) dynamic delivery system or the Amaxa nucleofection system (P3 primary kit, cat. no. V4XP-3024) according to the manufacturers’ instructions. For 0.5 × 105 HEK293FT cells, 100 pmol of labeled duplexed gRNA was mixed with 100 pmol of Cas9 protein (Alt-R S.p. Cas9 Nuclease 3NLS, cat. no. 1074182; IDT) in IDT nuclease-free duplex buffer and assembled for 30 minutes at room temperature. Afterwards, the CRISPR/Cas9-gRNA RNP was mixed with either Opti-MEM I reduced-serum medium and TransIT-X2 transfection reagent (HEK293FT) or with electroporation mix for the Amaxa nucleofection system according to the manufacturer’s protocol (Jurkat, and human iPSCs and CD34+ HSPCs, respectively). Jurkat cells (1.0 × 106) were electroporated with 300 pmol labeled duplexed gRNA mixed with 300 pmol Cas9 protein. Human iPSCs and CD34+ HSPCs (1.0 × 106) were electroporated with 400 pmol labeled duplexed gRNA and 400 pmol Cas9 protein. Transfection of HEK293FT cells with CX-rhodamine–labeled pMAX GFP plasmid was performed using TransIT-LT1 transfection reagent (cat. no. MIR2304; Mirus).
Genomic DNA isolation, PCR, Sanger sequencing and TIDE assay
Genomic DNA (gDNA) was isolated using the QIAamp DNA Mini Kit (cat. no. 51306; Qiagen) according to the manufacturer’s instructions. Polymerase chain reaction (PCR) with isolated gDNA and GADD45B-specific primers (forward: 5′-GACTACCGTTGGTTTCCGCAAC-3′, reverse: 5′-ATACATCAGGATACGGCAGCCC-3′) was carried out using the GoTaq Hot Start Polymerase Kit (cat. no. M5006; Promega) using 50 ng of gDNA template for each PCR reaction. PCR product purification was conducted with ExoSAP (ratio 3:1), a master mix of 1 part Exonuclease I 20 U/µL (cat. no. EN0581; Thermo Fisher Scientific) and 2 parts of FastAP thermosensitive alkaline phosphatase 1U/µL (cat. no. EF0651; Thermo Fisher Scientific). Sanger sequencing of purified PCR product was performed by Eurofins Genomics and analyzed using the TIDE (Tracking of Indels by Decomposition) webtool developed by Brinkman et al.22
Establishment of gene-edited cell lines and human iPSCs from single-cell clones using limiting dilution
Cells were serially diluted to 0.5 cells per 100 µL by adding of 60 cells in 12 mL Dulbecco’s modified Eagle medium or RPMI medium and pipetting of 100 µL of cell suspension per well of the 96-well plate. The 96-well plate was incubated for 2 to 3 weeks until appearance of growing cells.
Human iPSCs (15 000) were plated on a Geltrex-coated 10-cm dish in StemFlex medium (cat. no. A3349401; Thermo Fisher Scientific) and RevitaCell supplement (cat. no. A2644501; Thermo Fisher Scientific). Medium was changed every 24 hours without RevitaCell supplement. After 9 to 12 days, each colony was picked and transferred on the Geltrex-coated 96-well plate.
Cloning of the PCR products for the evaluation of the gene modification mode in GADD45B-edited clones
gDNA was isolated from gene-edited GADD45B+/− and GADD45B−/− iPSCs. The Cas9 RNP-targeted region of the GADD45B gene was amplified from gDNA using PCR with followed primers: forward 5′-GACTACCGTTGGTTTCCGCAAC-3′, reverse 5′-ATACATCAGGA TACGGCAGCCC-3′. PCR product was purified from the agarose gel using QIAquick Gel Extraction kit (cat no./ID: 28706; Qiagen) and cloned into the linearized pMiniT 2.0 vector using the NEB PCR Cloning Kit (cat. no. E1202S; New England Biolabs) followed by transformation of competent Escherichia coli and subsequent colony PCR of E coli colonies, according to the manufacturer’s instructions (cat. no. M5006; Promega). PCR products were analyzed using Sanger sequencing.
UV exposure and cell viability assay
Cells were irradiated with UV light (7 mJ/cm2) for 5 minutes and subsequently incubated for 2 hours under standard culture conditions before measuring the percentage of live GADD45B-targeting CRISPR/Cas9-gRNA RNP-transfected cells by quantitation of CX-rhodamine– or fluorescein-positive cells using a BD FACSCanto II flow cytometer.
Intracellular staining and fluorescence-activated cell sorter analysis of γH2AX (pSer139) protein
Intracellular γH2AX (pSer139) protein levels were measured in UV-irradiated cells. Briefly, cells were washed with phosphate-buffered saline and stained using the IntraSure kit (cat. no. 641778; BD) according to the manufacturer’s instructions and incubated with Alexa-Fluor 488 mouse anti-H2AX pSer139 antibody (1:100; cat. no. 560445; BD) for 15 minutes at room temperature, washed twice, fixed with 0.5% paraformaldehyde and analyzed using a FACSCanto II flow cytometer.
LORD-Q–DNA damage quantification
gDNA was isolated using a QIAamp DNA Mini Kit according to the manufacturer’s instructions. Long-run real-time PCR-based DNA-damage quantification (LORD-Q) was performed and analyzed according to the protocol of Lehle et al.23
Results
Design of the CRISPR/Cas9–gRNA RNP fluorescent labeling
We generated gRNA by annealing crRNA with tracrRNA. gRNA was covalently labeled with CX-rhodamine or fluorescein and incubated with recombinant Cas9 protein to generate CRISPR/Cas9–gRNA RNP complexes (Figure 1A). To assess the efficiency of fluorescent labeling, we transfected HEK293FT cells with a CX-rhodamine–labeled plasmid encoding GFP protein. GFP signals were colocalized with CX-rhodamine signals, thus proving efficient labeling of the GFP plasmid with CX-rhodamine (supplemental Figure 1A). An Agilent Bioanalyzer was used to further confirm that fluorescent labeling does not affect gRNA integrity (Figure 1B).
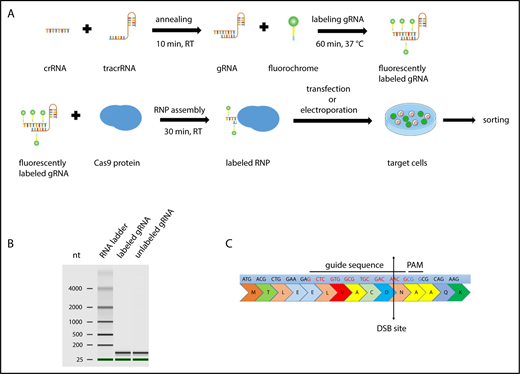
Scheme of CRISPR/Cas9–gRNA RNP labeling and cell transfection. (A) crRNA and tracrRNA were annealed at room temperature for 10 minutes. The resulting gRNA was labeled with fluorescein- or CX-rhodamine–coupled Label IT Tracker labeling reagent. The fluorescent GADD45B-targeting gRNA was assembled with recombinant Cas9 protein prior to transfection to assemble an active CRISPR/Cas9–gRNA RNP complex targeting human GADD45B. Cells were transfected with TransIT-X2 Transfection Reagent or by using the Amaxa Nucleofector System and were incubated for 24 hours before sorting the CX-rhodamine+ or fluorescein+ cells using a BD FACSAria II. After sorting, some of the cells were used for a single-cell culture, and the rest were used for DNA isolation or cell-based assays. (B) Virtual gel of an Agilent Bioanalyzer analysis revealing no difference in the size or quality of labeled gRNA compared with unlabeled gRNA. (C) GADD45B was targeted using gRNA (highlighted in red), which inserts a double-strand break at NM_015675.3 exon 1, 31 bp after ATG; NP_056490.2, p.N11.
Scheme of CRISPR/Cas9–gRNA RNP labeling and cell transfection. (A) crRNA and tracrRNA were annealed at room temperature for 10 minutes. The resulting gRNA was labeled with fluorescein- or CX-rhodamine–coupled Label IT Tracker labeling reagent. The fluorescent GADD45B-targeting gRNA was assembled with recombinant Cas9 protein prior to transfection to assemble an active CRISPR/Cas9–gRNA RNP complex targeting human GADD45B. Cells were transfected with TransIT-X2 Transfection Reagent or by using the Amaxa Nucleofector System and were incubated for 24 hours before sorting the CX-rhodamine+ or fluorescein+ cells using a BD FACSAria II. After sorting, some of the cells were used for a single-cell culture, and the rest were used for DNA isolation or cell-based assays. (B) Virtual gel of an Agilent Bioanalyzer analysis revealing no difference in the size or quality of labeled gRNA compared with unlabeled gRNA. (C) GADD45B was targeted using gRNA (highlighted in red), which inserts a double-strand break at NM_015675.3 exon 1, 31 bp after ATG; NP_056490.2, p.N11.
Specific knockout of GADD45B using labeled CRISPR/Cas9–gRNA RNP
To functionally validate the knockout of weakly expressed genes with inducible mRNA expression using labeled CRISPR/Cas9–gRNA RNP, we chose to disrupt the human growth arrest and DNA-damage-inducible 45 β (GADD45B) gene.24 We designed crRNA for exon 1 of GADD45B (Figure 1C), generated labeled GADD45B CRISPR/Cas9–gRNA RNP, and transfected HEK293FT cells, the Jurkat T-ALL cell line, bone marrow CD34+ HSPCs, and iPSCs. We detected CX-rhodamine or fluorescein signals 6 hours (HEK293FT cells) or 12 hours (Jurkat cells, CD34+ HSPCs, and iPSCs) after transfection. Transfection efficiency varied between 40% and 80%, depending on the cell type (Figure 2A-B). The intracellular fluorescent signal disappeared ∼48 hours after transfection. Labeling did not affect the gene-editing efficiency of CRISPR/Cas9–gRNA RNP, as assessed by Sanger sequencing and tracking of indels by decomposition (TIDE) assay analysis of HEK293FT cells, Jurkat cells, CD34+ HSPCs, and human iPSCs transfected with labeled or unlabeled GADD45B-targeting CRISPR/Cas9–gRNA RNP (Figure 2C). Using fluorescein or rhodamine signals of labeled CRISPR/Cas9–gRNA RNP, we sorted and enriched gene-edited fluorescent cells by flow cytometry. Gene-modification efficiency in sorted cells was approximately 40% in iPSCs, 60% in HSPCs, and 70% in Jurkat cells (Figure 2D).
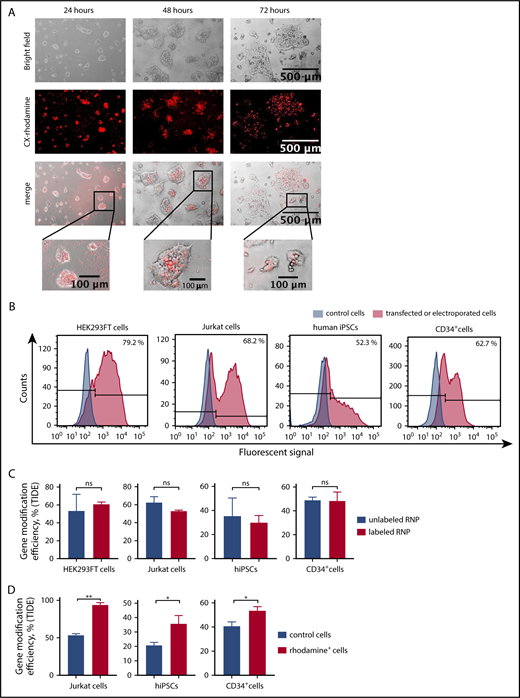
Transfection- and genome-editing efficiency in different cell types using CX-rhodamine–labeled CRISPR/Cas9–gRNA RNP targeting GADD45B. (A) HEK293FT cells were transfected with CX-rhodamine–labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. Fluorescence signal could be detected for up to 72 hours posttransfection. Representative images of 3 experiments are shown. (B) HEK293FT cells, Jurkat cells, human iPSCs, and CD34+ cells were transfected with labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. At 24 hours posttransfection, cells were harvested and measured for transfection efficiency using a BD FACSCanto II flow cytometer. Representative line graphs of 3 independent experiments are shown. (C) HEK293FT cells, Jurkat cells, human iPSCs, and CD34+ HSPCs were transfected with unlabeled or labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP and analyzed for gene-modification efficiency using a TIDE assay. (D) Jurkat cells, human iPSCs, and CD34+ HSPCs were transfected with CX-rhodamine–labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP and sorted 24 hours posttransfection using a flow cytometer. Genomic DNA was isolated 48 hours posttransfection from the total population of transfected cells and from sorted CX-rhodamine+ or fluorescein+ cells. TIDE assay analysis showed significantly higher gene modification efficiency in CX-rhodamine+ cells. Data in panels C and D are mean ± standard deviations derived from 3 (HEK293FT cells, Jurkat cells, CD34+ HSPCs) or 4 (iPSCs) independent experiments. *P ≤ .05, **P ≤ .01, Student t test. ns, not significant.
Transfection- and genome-editing efficiency in different cell types using CX-rhodamine–labeled CRISPR/Cas9–gRNA RNP targeting GADD45B. (A) HEK293FT cells were transfected with CX-rhodamine–labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. Fluorescence signal could be detected for up to 72 hours posttransfection. Representative images of 3 experiments are shown. (B) HEK293FT cells, Jurkat cells, human iPSCs, and CD34+ cells were transfected with labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. At 24 hours posttransfection, cells were harvested and measured for transfection efficiency using a BD FACSCanto II flow cytometer. Representative line graphs of 3 independent experiments are shown. (C) HEK293FT cells, Jurkat cells, human iPSCs, and CD34+ HSPCs were transfected with unlabeled or labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP and analyzed for gene-modification efficiency using a TIDE assay. (D) Jurkat cells, human iPSCs, and CD34+ HSPCs were transfected with CX-rhodamine–labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP and sorted 24 hours posttransfection using a flow cytometer. Genomic DNA was isolated 48 hours posttransfection from the total population of transfected cells and from sorted CX-rhodamine+ or fluorescein+ cells. TIDE assay analysis showed significantly higher gene modification efficiency in CX-rhodamine+ cells. Data in panels C and D are mean ± standard deviations derived from 3 (HEK293FT cells, Jurkat cells, CD34+ HSPCs) or 4 (iPSCs) independent experiments. *P ≤ .05, **P ≤ .01, Student t test. ns, not significant.
Transfection of cells with a nontargeting RNP, consisting of tracrRNA and Casp9 alone, did not affect genome integrity (supplemental Figure 1B). We also compared fluorescent labeling of crRNA with the expression of Cas9–EGFP fusion protein. We detected much lower editing efficiency of the fused Cas9-EGFP protein assembled with GADD45B-targeting gRNA compared with CRISPR/Cas9–gRNA RNP (supplemental Figure 2A).
Cloning of the PCR products from genomic DNA of single-cell clones of the gene-edited Jurkat cells and iPSCs revealed compound heterozygous GADD45B frameshift mutations in Jurkat cells (supplemental Figure 3A), and heterozygous, as well as homozygous, GADD45B deletions in iPSC clones (supplemental Figure 3B). We found no off-target activities of the GADD45B-specific crRNA, with the possibility of 3 bp mismatches. We also detected only a small number of potential off-target sites in other genes that could be targeted (<3 mismatches) with low probability (0.2%-0.9%) by the GADD45B-specific crRNA (supplemental Table 1). However, we did not detect any mutations in the selected gene regions in the edited cell types used in our study (supplemental Figure 4A).
GADD45B is essential for the induction of DNA damage response in human hematopoietic cells and iPSCs
We further performed functional studies of the effect of GADD45B knockout on cell growth and sensitivity to UV-induced DNA damage. Remarkably, we detected a strongly diminished viability of GADD45B-deficient HEK293FT cells, Jurkat cells, iPSCs, and CD34+ HSPCs compared with control transfected cells (Figure 3A). We also found markedly elevated susceptibility of GADD45B-deficient Jurkat cells and CD34+ HSPCs to UV-induced DNA damage, as documented by increased expression of the DNA damage marker γH2AX (phospho-Ser139). Basal levels of γH2AX (phospho-Ser139) were also elevated in GADD45B-modified HSPCs and Jurkat cells (Figure 3B-C). In addition, we detected an accumulation of DNA lesions in GADD45B-deficient cells compared with wild-type Jurkat cells (Figure 3D). As revealed by LORD-Q DNA damage–quantification analysis,23 GADD45B-deficient cells exhibited increased DNA damage rates in the mitochondrial DNA (mtDNA), as well as in 2 analyzed genomic loci of nuclear DNA. Based on these observations, we conclude that GADD45B knockout in cells transfected with labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP led to increased susceptibility to DNA damage.
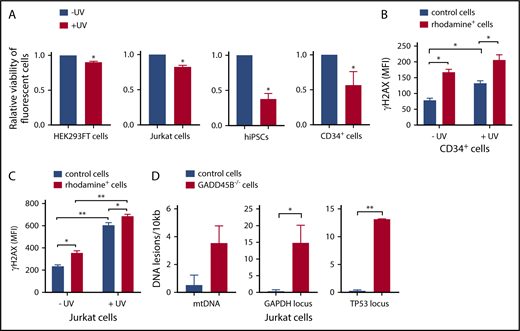
GADD45B knockout leads to reduced cell viability and increased UV-induced cellular stress. (A) Cell viability of HEK293FT cells, Jurkat cells, iPSCs, and CD34+ HSPCs, transfected with labeled tracr-Cas9 RNP (nontarget RNP) or with labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP, was measured after exposing the cells to UV for 5 minutes, followed by 2 hours of additional incubation. Relative viability of nonirradiated control cells was set as 1.0. (B) CD34+ HSPCs were transfected with fluorescein-labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. After 48 hours, the cells were exposed to UV irradiation for 5 minutes. Following 2 hours of further incubation, intracellular γH2AX (phospho-Ser139) levels were measured by flow cytometry. (C) Jurkat cells were transfected with CX-rhodamine–labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. After 48 hours, the total population was exposed to UV irradiation for 5 minutes, followed by 2 hours of incubation before performing intracellular staining and FACS analysis for the DNA damage marker γH2AX (phospho-Ser139). (D) mtDNA damage (left panel) and nuclear DNA damage in the GAPDH locus (middle panel) and TP53 locus (right panel) were quantified in Jurkat control cells and a GADD45B−/− Jurkat clone using the LORD-Q method. Data are mean ± standard deviation from 3 (A-B) or 2 (C-D) independent experiments, each performed in duplicates. *P ≤ .05, **P ≤ .01, Student t test.
GADD45B knockout leads to reduced cell viability and increased UV-induced cellular stress. (A) Cell viability of HEK293FT cells, Jurkat cells, iPSCs, and CD34+ HSPCs, transfected with labeled tracr-Cas9 RNP (nontarget RNP) or with labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP, was measured after exposing the cells to UV for 5 minutes, followed by 2 hours of additional incubation. Relative viability of nonirradiated control cells was set as 1.0. (B) CD34+ HSPCs were transfected with fluorescein-labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. After 48 hours, the cells were exposed to UV irradiation for 5 minutes. Following 2 hours of further incubation, intracellular γH2AX (phospho-Ser139) levels were measured by flow cytometry. (C) Jurkat cells were transfected with CX-rhodamine–labeled GADD45B-targeting CRISPR/Cas9–gRNA RNP. After 48 hours, the total population was exposed to UV irradiation for 5 minutes, followed by 2 hours of incubation before performing intracellular staining and FACS analysis for the DNA damage marker γH2AX (phospho-Ser139). (D) mtDNA damage (left panel) and nuclear DNA damage in the GAPDH locus (middle panel) and TP53 locus (right panel) were quantified in Jurkat control cells and a GADD45B−/− Jurkat clone using the LORD-Q method. Data are mean ± standard deviation from 3 (A-B) or 2 (C-D) independent experiments, each performed in duplicates. *P ≤ .05, **P ≤ .01, Student t test.
GADD45B protects iPSCs from UV stress in a dose-dependent manner
Interestingly, GADD45B+/− and GADD45B−/− iPSCs retained pluripotency (Figure 4A), but we detected markedly elevated phospho-Ser139 γH2AX levels in GADD45B-haploinsufficient and GADD45B-homozygous–knockout iPSCs. Elevated DNA damage was observed under steady-state conditions and, more profoundly, upon genotoxic UV exposure compared with wild-type iPSCs (Figure 4B). In line with upregulated γH2AX (phospho-Ser139) levels, we measured elevated DNA damage in GADD45B+/− and GADD45B−/− iPSCs, as determined by the LORD-Q DNA damage–quantification assay (Figure 4C-D). These data are in accordance with our observations in GADD45B-deficient Jurkat cells and HSPCs.
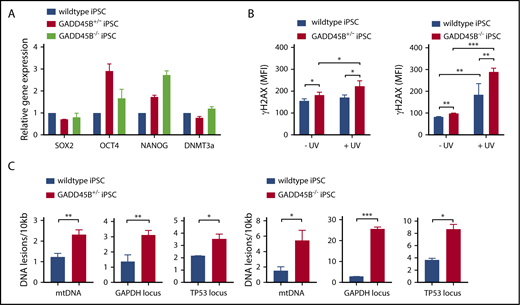
Heterozygous and homozygous GADD45B knockout in human iPSCs results in high levels of DNA damage. (A) Pluripotency state of GADD45B+/− and GADD45B−/− iPSCs was assessed by real-time quantitative PCR and compared with validated healthy donor–derived human iPSCs expressing wild-type GADD45B. (B) GADD45B wild-type, GADD45B+/−, and GADD45B−/− iPSCs were irradiated with UV light for 5 minutes, incubated under cell culture conditions for 2 hours, and stained for intracellular γH2AX (phospho-Ser139). DNA damage in GADD45B wild-type and GADD45B heterozygous-knockout (C) or homozygous-knockout (D) iPSCs was quantified by the LORD-Q method. Cells were analyzed for mtDNA damage and nuclear DNA damage in the GAPDH and TP53 gene loci. All data are mean ± standard deviation derived from 3 independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, Student t test.
Heterozygous and homozygous GADD45B knockout in human iPSCs results in high levels of DNA damage. (A) Pluripotency state of GADD45B+/− and GADD45B−/− iPSCs was assessed by real-time quantitative PCR and compared with validated healthy donor–derived human iPSCs expressing wild-type GADD45B. (B) GADD45B wild-type, GADD45B+/−, and GADD45B−/− iPSCs were irradiated with UV light for 5 minutes, incubated under cell culture conditions for 2 hours, and stained for intracellular γH2AX (phospho-Ser139). DNA damage in GADD45B wild-type and GADD45B heterozygous-knockout (C) or homozygous-knockout (D) iPSCs was quantified by the LORD-Q method. Cells were analyzed for mtDNA damage and nuclear DNA damage in the GAPDH and TP53 gene loci. All data are mean ± standard deviation derived from 3 independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, Student t test.
Discussion
In the present study, we have developed a new method of CRISPR/Cas9-mediated gene editing in primary HSPCs and iPSCs using fluorescent labeling of CRISPR/Cas9–gRNA RNP complexes. Using fluorescein or CX-rhodamine signals from labeled CRISPR/Cas9–gRNA RNP, we were able to enrich gene-edited cells by fluorescent cell sorting. For clinical settings, it is essential to select and enrich gene-edited HSPCs because of the limited capacity of these cells to divide, to retain their engraftment capability, and to generate unlimited numbers of progeny cells. Sorting of the labeled cells also allows removal of untargeted HSPCs that may compete with gene-edited cells. In addition, application of CRISPR/Cas9 RNP decreases the probability and frequency of off-target effects, because CRISPR/Cas9 RNP activity is preserved in cells for only ∼48 hours.
Dever et al recently reported the CRISPR/Cas9 RNP–based modification and enrichment of human HSPCs by introduction of a repair template targeting the human β-globin gene through a GFP-expressing adeno-associated virus–based vector.3 Although considered safer than retroviral constructs, adeno-associated virus–based expression constructs may induce antiviral host immune responses and may integrate into the host genome nonspecifically. Because RNP-mediated gene knockout allows the efficient virus- and DNA-free transfection and selection of edited cells, future studies should further investigate our method of fluorescent labeling of CRISPR/Cas9 RNP in a gene-correction approach mediated by homology-directed repair, which additionally requires a donor template DNA. Again, the short exposure of cells to CRISPR/Cas9 RNP activity makes it superior to virus-based delivery techniques.
We also tested EGFP-conjugated CRISPR Cas9 RNP but found that EGFP tagging resulted in reduced editing efficiency compared with unlabeled CRISPR Cas9 RNP. At the same time, labeling of CRIPSR/Cas9 RNP using our method described here does not affect editing.
Although we studied gene editing in primary hematopoietic stem cells and iPSCs, the method described here may be extended to other primary cell types. Gene-modification efficiency is dependent on the cell type, cell cycle stage, activation of DNA-repair pathways, chromatin dynamics at the gRNA-targeted gene locus, and the delivery method.25,25-27 Delivery and editing protocols may be further improved (eg, by synchronizing of the targeted cells), which could increase nuclear uptake of the RNP components and chromatin relaxation.
For a proof of principle, we chose to target the GADD45B gene and found that homo- and even heterozygous deficiency in GADD45B led to increased susceptibility to DNA damage. GADD45B belongs to a family of evolutionarily conserved GADD45 proteins28 that functions as stress sensors regulating cell cycle, survival, and apoptosis in response to various stress stimuli.29 With some degree of redundancy, GADD45 proteins exhibit specific functions, depending on the stimulus and cell type. Gadd45b-knockout mice are viable, but Gadd45b−/− primary mouse embryonic fibroblasts proliferate slowly and accumulate increased levels of DNA damage and features of premature senescence.30-33 It has also been shown that myeloid differentiation is compromised in Gadd45b−/− mice.33 Nothing was known about GADD45b functions in the DNA damage response of human HSPCs and iPSCs. Our study demonstrates an essential role for GADD45B in the survival and protection from DNA damage in human leukemia cells and CD34+ HSPCs that are not compensated for by other GADD45 proteins.
Our study is the first report describing transfection of iPSCs with labeled CRISPR/Cas9–gRNA RNP with high transfection and knockout efficiency. In future studies, gene correction using labeled RNP complexes for ex vivo gene therapy and transplantation could be tested in CD34+ cells. There are also reports describing in vivo gene correction in neurons, hepatocytes, and cardiomyocytes.3,14-20 Currently, there are many challenges associated with using CRISPR/Cas9 approaches. Efficient gene editing is reliant on the successful delivery of the Cas9 nuclease and the gRNA, which is especially cumbersome in primary cell types resistant to DNA transfection. Moreover, plasmid and viral delivery lead to persistent overexpression of Cas9, which can potentially result in off-targets. In contrast, direct delivery of RNP complexes, which are gradually cleared by intracellular degradation over time, does not result in Cas9 persistence and, therefore, reduces potential off-target effects. Furthermore, we did not detect toxic or inhibitory effects of the labeling on the gene-editing efficiency or on cell growth. As a result of their short exposure, it is also unlikely that the fluorochrome dyes used in our study are immunogenic in vivo. However, further optimizations are required for the in vivo application of our method. For instance, different dyes should be tested for their immunogenicity.
Taken together, chemical labeling of the gRNA and the direct transfection of RNP complexes provide a simple, safe, and efficient method that could considerably expand future therapeutic avenues for CRISPR/Cas9-mediated gene editing.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Regine Bernhardt, Ingeborg Steiert, Karin Haehnel, and Ursula Hermanutz-Klein for the excellent technical assistance. The authors thank the FACS Core Facility of the University Hospital Tübingen, especially Kristin Bieber, Cornelia Grimmel, and Stella Autenrieth, for assistance with cell sorting. They also thank Kai Witte and D. Campana for providing 8E permeabilization reagent.
This work was supported by the Excellence Initiative of the Faculty of Medicine, University of Tübingen (J.S.), Madeleine Schickedanz Kinderkrebsstiftung (M.N.), German Cancer Consortium (P.M. and J.S.), DFG (M.K.), the Fresenius Foundation (M.K.), the Fritz Thyssen Foundation (B.D.), and the Jose Carreras Leukemia Foundation (J.S. and B.D.).
Authorship
Contribution: M.N., P.M., and J.S. made initial observations, designed the experiments, analyzed the data, and wrote the manuscript; M.N. and P.M. performed labeling of CRISPR/Cas9–gRNA RNP complexes, electroporation or transfection of cells, cell culture, treatment with UV light, FACS analysis, cell counts, cell sorting, Sanger sequencing, and LORD-Q experiments; B.D. and M.N. conducted experiments with iPSCs; M.N. and T.S. established transfection of iPSCs; Y.X. performed real-time quantitative PCR for pluripotency markers; D.A. performed image analysis of HEK293FT cells; K.S.-O. assisted with LORD-Q experiments and provided insightful comments; and M.K. and K.W. assisted with the interpretation of data and provided insightful comments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Julia Skokowa, Division of Translational Oncology, Department of Hematology, Oncology, Clinical Immunology, Rheumatology and Pulmonology, University Hospital Tübingen, Otfried-Müller-Str 10, 72076 Tübingen, Germany; e-mail: julia.skokowa@med.uni-tuebingen.de.

