

Key Points
Deletion of SNAP23 results in complete loss of platelet granule secretion.
Loss of SNAP23 leads to macrothrombocytopenia and severely reduced thrombosis and hemostasis.

Abstract
Platelet secretion is central to physiological and pathophysiological platelet function. SNAP23 has long been implicated as being a principal SNARE protein regulating platelet granule secretion, although this has not been definitively demonstrated in genetic models. Here, using a platelet-specific conditional SNAP23 knockout mouse, we show that absence of SNAP23 results in complete ablation of dense granule, α granule, and lysosomal secretion. Measured granule cargo content and granule numbers were normal, suggesting SNAP23 regulates fusion of granules with the extracellular membrane, rather than granule loading or formation. A macrothrombocytopenia was also observed, which, combined with ablation of secretion, resulted in a pronounced bleeding defect in a tail bleed assay and almost complete ablation of arterial and venous thrombosis. The macrothrombocytopenia was not due to reduced megakaryopoiesis but instead likely was due to the increased loss of platelets through bleeding, consistent with an increase in platelet total RNA content indicating a greater number of reticulated platelets. The data definitively show SNAP23 to be critical for granule release of any kind from platelets, irrespective of stimulus, and this is the first single gene to be shown to be universally essential for exocytosis in platelets.
Introduction
Platelet granule secretion supports not only thrombosis and hemostasis but also a range of other physiological and pathophysiological processes of clinical relevance, including inflammation, tissue healing and repair, neovascularization, and cancer metastasis and progression.1 Bleeding disorders associated with granule secretion defects fall into 2 categories: (1) granule biogenesis defects (eg, gray platelet syndrome2 ) and (2) secretion machinery defects (eg, familial hemophagocytic lymphohistiocytosis [FHL]3 ). Several key components of the platelet secretion machinery have been identified in recent years, notably Syntaxin 114 and Munc18b,5 which were shown in FHL patients to have negligible dense (δ)- and alpha (α)-granule secretion, but with some lysosomal release. Munc13-4 is also absolutely required for δ-granule release and plays a major role in α-granule and lysosomal secretion,6 although part of the deficit in α-granule secretion can be attributed to the loss of adenosine 5′-diphosphate (ADP) secretion and subsequent paracrine ADP signaling.7,8
SNAP23 has long been recognized as an important regulator of platelet granule release,9-11 although to date there have been no studies using a genetic approach to determine precisely its role. Recently, the report that SNAP29, the other SNAP family member readily detectible in platelets, has a very limited role in platelet granule secretion12 supported the idea that SNAP23 may be the key Qbc SNARE in platelets. Here, we use a platelet-specific conditionally targeted SNAP23 null mouse model, showing for the first time that the gene is critical for all platelet granule secretion. This has important implications for our understanding of the molecular regulation of platelet secretion, but also provides a novel way to assess the role of that secretion in hemostatic and nonhemostatic processes, through single-gene targeting.
Methods
Mice
Snap23tm1a(EUCOMM)Wtsi mice were from the Wellcome Trust Sanger Institute. Mice were crossed with mLacbFlpe mice globally expressing Flp recombinase, with subsequent crossing with platelet factor 4 (PF4)–Cre mice. Mice were bred and maintained at the University of Bristol in accordance with United Kingdom Home Office regulations. All procedures were approved by the United Kingdom Home Office in accordance with the Animals (Scientific Procedures) Act of 1986 (PPL 30/3445).
Platelet preparation
For most experiments, blood was drawn into 4% citrate and processed to washed platelets as previously described.12 Platelets were used at assay-specific concentrations in the presence of 10 µM indomethacin and 0.02 units/mL apyrase.
For calcium signaling, blood was drawn into acid citrate dextrose (71 mM citric acid, 85 mM sodium citrate, 111 mM glucose), diluted with saline, and centrifuged at 200g for 5 minutes in the presence of 140 nM prostaglandin E1 and 0.5 units/mL apyrase. Platelet-rich plasma was harvested, diluted with saline, and centrifuged at 700g for 10 minutes in the presence of 5 µM EDTA. The pellet was resuspended in CGS buffer (0.38% citrate, 0.54% d-glucose, 120 mM NaCl, pH 6.5) and then centrifuged at 700g for 10 minutes. The pellet was resuspended to 400 × 109/L in CGS buffer (0.38% citrate, 0.54% d-glucose, 120 mM NaCl, pH 6.5) for use in calcium signaling assay.
Hematology
Citrated blood was sampled on a Horiba Pentra ES60 analyzer and corrected for draw volume.
Adenosine triphosphate (ATP) secretion assay
Platelets at 200 × 109/L were incubated with 1/100 Chronolume luciferin luciferase reagent (Laboratory Medics, Abingdon, United Kingdom) prior to agonist stimulation at 37°C under nonstirring conditions in a Tecan Infinite M200Pro plate reader. Samples were standardized with the injection of 1 nmol of ATP.
Flow cytometry
For surface receptor analysis, platelets at 20 × 109/L were stimulated for 10 minutes under nonstirring conditions in the presence of PE-Jon/A and fluorescein isothiocyanate (FITC)–P-selectin as previously described.12 FITC–anti-CD41/αIIb, α2, glycoprotein VI (GPVI), and glycoprotein Ib-alpha (GPIb-α) were from Emfret Analytics (Eibelstadt, Germany). FITC–anti-PECAM (CD31) was from eBioscience (Thermo Fisher Scientific, United Kingdom).
For total RNA determination, platelets were incubated under nonstirring conditions with 10 µg/mL Hoechst 33342 (Molecular Probes, Thermo Fisher Scientific) for 45 minutes at 30°C to saturate DNA, prior to incubating with 0.5 µg/mL Pyronin Y (Acros Organics, Thermo Fisher Scientific) for 15 minutes at 30°C. Unstained, Hoechst 33342-only, and Pyronin Y-only samples were run as controls. Samples were diluted prior to immediate analysis.
All samples were analyzed on an Accuri C6 Plus flow cytometer (BD Biosciences, Oxford, United Kingdom).
Platelet aggregometry
Washed platelets at 200 × 109/L were stimulated under stirring conditions (1000 rpm) in a Chrono-Log Model 700 aggregometer (Laboratory Medics), with data acquired and analyzed using Aggrolink 8 software.
Immunoblotting
Platelets at 200 × 109/L were blotted as previously described.12 Anti-SNAP23, SNAP29, syntaxin 11, VAMP8, and munc18b were from Synaptic Systems (Goettingen, Germany); anti-PECAM, Talin, CD62-P, and munc13-4 were from Insight Biotechnology (Wembley, United Kingdom), and anti-Syntaxin 8 was from Bio-Techne Ltd (Abingdon, United Kingdom). Anti-VAMP7, pSykY525/526, total Syk, pSTAT5Y694, and pJAKY1007/1008 were from New England Biolabs (Hitchin, United Kingdom), and anti-STXBP5 was from BD Biosciences (Wokingham, United Kingdom). Species-appropriate near-infrared (700/800 nm) secondary antibodies were from Jackson Immunoresearch (Stratech, Ely, United Kingdom). Membranes were imaged on a LI-COR Odyssey CLx (LI-COR, Cambridge, United Kingdom).
CXCL4/PF4 enzyme-linked immunosorbent assay (ELISA) and β-hexosaminidase release assay
Platelets at 200 × 109/L were stimulated for 10 minutes at 37°C under nonstirring conditions. Samples were centrifuged 15 seconds at 12 000g to pellet the cells. The supernatants were removed and spun again under the same conditions, before being snap-frozen and stored at −80°C until required. Total samples were generated by lysing unstimulated platelets with an equal volume of 1% Triton X-100 (TX100) in Tyrode's buffer, before being snap-frozen.
For CXCL4/PF4 ELISA, stimulated samples were diluted 1/250 (totals 1/500) and used as per manufacturer’s instructions (Mouse CXCL4/DuoSet; R&D Systems, Abingdon, United Kingdom).12
For β-hexosaminidase release assay, an equal volume of releasate was added to 20 µL 5 mM 4-nitrophenyl n-acetyl-β-d-glucosaminide (Sigma, Poole, United Kingdom) substrate in citrate-phosphate buffer (0.2 M sodium hydrogen phosphate, 0.1 M citric acid, pH 4.2) and incubated for 1 hour at 37°C. Absorbance was measured immediately at 405 nm.13
Plates were read on a Tecan Infinite M200Pro microplate reader.
Calcium signaling
Platelets were incubated for 45 minutes at 37°C with 0.1% Pluronic F127 and 4 µM Fura-PE3 (Thermo Fisher Scientific) then centrifuged at 700g for 10 minutes and resuspended in HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid)–Tyrode's buffer with 1 mM MgCl2 and 1 mM CaCl2. Platelets were rested for 30 minutes and then diluted to 100 × 109/L and stimulated at 37°C under nonstirring conditions on a Tecan Infinite M200Pro plate reader. Samples were lysed with 0.2% TX100 for total calcium content.
Transmission electron microscopy (TEM)
Platelet-rich plasma was prepared for TEM as described previously.12 Sections were imaged on a Philips CM100 equipped with a side-mount MegaView III camera (Olympus Soft Imaging Solutions). For granule analysis, total platelet section area was measured and α- and δ-granule numbers counted in at least 10 randomly selected fields of view using ImageJ 1.46.
Bone marrow histology
Femurs from 8- to 10-week-old mice were excised and fixed in 4% paraformaldehyde, before being decalcified in 20% to 30% formic acid for 2 weeks, stained with hematoxylin and eosin, and sectioned. Sections were imaged on a Leica DM IRB wide-field microscope with a Leica DFC 450C camera. For megakaryocyte counting, 10 randomly selected fields of view were imaged using a ×20 objective, with the total marrow area measured and megakaryocyte number counted using ImageJ 1.46.
Tail bleeding assay
Eight- to 10-week-old male or female mice were anesthetized by intraperitoneal injection of a 100 mg/kg ketamine/10 mg/mL xylazine mix. The tail tip was resected 5 mm by scalpel and immediately immersed in prewarmed saline (37°C). Time until cessation of bleeding was recorded, up to a maximum of 10 minutes.14
Arterial thrombosis assay
Male or female mice were anesthetized by intraperitoneal injection of a 100 mg/kg ketamine/10 mg/mL xylazine mix. Platelets were labeled with an IV administration of 0.1 µg/g body weight Dylight-488–conjugated anti-GPIbβ antibody (Emfret Analytics) into the jugular vein. Carotid arteries were surgically exposed, and a 2 × 1-mm piece of filter paper saturated with 24% ferric chloride was applied for 1 minute. Thrombus formation was followed for 10 minutes by fluorescence time-lapsed microscopy using an Olympus BX51-WI microscope with a Rolera XR digital camera and recorded using Streampix 4.24.0 (Norpix, Montreal, QC Canada). Images were analyzed using Image J 1.46 as previously described.7
Venous thrombosis assay
Deep vein thrombosis was induced in mice according to the procedure described by Payne et al,15 with modification. Briefly, adult mice were anesthetized with 5% isoflurane to induce general anesthesia and then 1% to maintain during surgery, which was carried out under aseptic conditions. Mice were placed in a supine position and subject to laparotomy. The inferior vena cava (IVC) was ligated just distal to the left renal vein using a fine (7-0) suture to completely block blood flow. Large branches of the IVC were also ligated to reduce variation in thrombus formation.16 Animals were given analgesic and placed on heated pads after recovery from surgery. Forty-eight hours after surgery, mice were euthanized and IVCs were dissected to assess thrombus formation and processed for histology.
IVC immunohistochemistry
Fresh tissues were quickly dissected and frozen in optimal cutting temperature compound and sectioned (5-μm thickness). Tissue sections were fixed with 4% formaldehyde/ phosphate-buffered saline for 30 minutes, permeabilized with 0.2% TX100 in phosphate-buffered saline for 30 minutes at room temperature, and probed with antibody against platelet marker CD41, leukocyte marker CD45 for overnight at 4°C. Appropriate secondary antibodies with Alexa-fluorophores were used for confocal microscopy.
Data presentation and statistical analysis
Data were analyzed using GraphPad Prism 7. Data are presented as mean ± standard error of the mean (SEM) and were analyzed by Student t test unless otherwise stated.
Results
SNAP23 has been implicated in platelet secretion,9-11 but genetic studies have not previously been performed because gene deletion causes early embryonic lethality in the ubiquitous knockout.17 We therefore generated a platelet-specific knockout mouse and confirmed SNAP23 as absent (Figure 1); however, a marked thrombocytopenia was unexpectedly observed in the platelet-specific knockout, with a 61% reduction in platelet count and 27% increase in mean platelet volume. White and red blood cell counts were normal (Table 1). Despite the macrothrombocytopenia, the expression of other platelet secretion regulators (Munc13-4, Munc18b, Syntaxin 8, Syntaxin 11, STXBP5, VAMP7, VAMP8) and the α-granule marker P-selectin (CD62P) show no significant changes (Figure 1).

Major secretion regulators are unchanged in the absence of SNAP23. (A) Unstimulated platelets at 200 × 109/L were lysed and proteins were run on sodium dodecyl sulfate–polyacrylamide gel electrophoresis, before transfer to polyvinylidene difluoride membrane (Immobilon FL), and probed as indicated. Membranes were imaged on a LiCor Odyssey CLx with species-appropriate near-infrared (700/800 nm)–conjugated secondary antibodies. Data are representative of at least 3 independent experiments. (B) Densitometric analysis of platelet secretion regulators or granule cargo proteins. Data are expressed as a percent of the wild-type (WT) average per blot. Data were compared by 2-way analysis of variance (ANOVA) with Sidak’s multiple comparisons test. No data were significant. Munc13-4, VAMP8; WT (n = 14), knockout (KO) (n = 11). CD62-P (P-selectin), Syntaxin 11, SNAP29; WT and KO (n = 8). Munc18b, STXBP5, VAMP7; WT (n = 5), KO (n = 4). PECAM, Syntaxin 8; WT (n = 5), KO (n = 3).
Major secretion regulators are unchanged in the absence of SNAP23. (A) Unstimulated platelets at 200 × 109/L were lysed and proteins were run on sodium dodecyl sulfate–polyacrylamide gel electrophoresis, before transfer to polyvinylidene difluoride membrane (Immobilon FL), and probed as indicated. Membranes were imaged on a LiCor Odyssey CLx with species-appropriate near-infrared (700/800 nm)–conjugated secondary antibodies. Data are representative of at least 3 independent experiments. (B) Densitometric analysis of platelet secretion regulators or granule cargo proteins. Data are expressed as a percent of the wild-type (WT) average per blot. Data were compared by 2-way analysis of variance (ANOVA) with Sidak’s multiple comparisons test. No data were significant. Munc13-4, VAMP8; WT (n = 14), knockout (KO) (n = 11). CD62-P (P-selectin), Syntaxin 11, SNAP29; WT and KO (n = 8). Munc18b, STXBP5, VAMP7; WT (n = 5), KO (n = 4). PECAM, Syntaxin 8; WT (n = 5), KO (n = 3).
Hematology
| Parameter | SNAP23 WT | SNAP23 KO | P | ||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | N | Mean | SEM | N | ||
| WBC, ×109/L | 7.1 | 0.4 | 28 | 7.6 | 0.5 | 22 | ns |
| RBC, ×1012/L | 10.3 | 0.3 | 28 | 10.7 | 0.2 | 22 | ns |
| PLT, ×109/L | 815.3 | 25.9 | 28 | 318.7 | 11.2 | 22 | <.0001 |
| MPV, µm3 | 5.5 | 0.0 | 28 | 7.0 | 0.1 | 22 | <.0001 |
| Parameter | SNAP23 WT | SNAP23 KO | P | ||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | N | Mean | SEM | N | ||
| WBC, ×109/L | 7.1 | 0.4 | 28 | 7.6 | 0.5 | 22 | ns |
| RBC, ×1012/L | 10.3 | 0.3 | 28 | 10.7 | 0.2 | 22 | ns |
| PLT, ×109/L | 815.3 | 25.9 | 28 | 318.7 | 11.2 | 22 | <.0001 |
| MPV, µm3 | 5.5 | 0.0 | 28 | 7.0 | 0.1 | 22 | <.0001 |
MPV, mean platelet volume; ns, not significant; PLT, platelet count; RBC, red blood cell count; WBC, white blood cell count.
Absence of SNAP23 led to complete ablation of δ-granule (as measured by ATP release; Figure 2A), α granule (as measured by PF4 release, Figure 2B; and P-selectin exposure, Figure 2C) and lysosome (as measured by β-hexosaminidase release, Figure 2D) secretion in response to thrombin or CRP, strikingly indicating a critical role for SNAP23 in secretion from all platelet granules. Total levels of PF4 and β-hexosaminidase activity in the SNAP23 knockout are equivalent to wild type (Figure 2Biii and Diii, respectively), indicating that absence of secretion was due to defective secretion machinery, and not a granule loading defect. Likewise, the defective α-granule P-selectin exposure in response to thrombin stimulation could not be rescued in the knockout by costimulating with ADP (Figure 2Ciii), suggesting that it is not the absence of paracrine ADP signaling causing the α-granule secretion defect.
![Figure 2. Secretion is ablated in the absence of SNAP23. (A) Washed platelets at 200 × 109/L were stimulated under nonstirring conditions at 37°C in a Tecan Infinite M200Pro plate reader in the presence of 1/100 Chronolume luciferin luciferase. Median traces of platelets stimulated (arrow) with 0.4 units/mL thrombin (i) and 20 µg/mL collagen-related peptide (CRP) (ii), respectively. (iii) Maximum ATP released upon stimulation (WT [n = 6-8], KO [n = 5]). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (B) Washed platelets at 200 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions at 37°C for 10 minutes before supernatant was harvested, run on a PF4 ELISA, and read at 450 nm. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 3-4), KO (n = 3). (iii) Lysed basal platelets were used to determine total platelet PF4 content (n = 6). (C) Washed platelets at 20 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions for 10 minutes at room temperature in the presence of FITC-conjugated anti–P-selectin. Curves were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 7-8), KO (n = 5). (iii) P-selectin binding at maximal thrombin (Thr; 0.4 units/mL) when costimulated with 10 µM ADP. WT (n = 3-7), KO (n = 4-5). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (D) Washed platelets at 200 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions at 37°C for 10 minutes before supernatant was harvested, run through a β-hexosaminidase colorimetric assay, and read at 405 nm. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 3-4), KO (n = 3). (iii) Lysed basal platelets were used to determine total platelet β-hexosaminidase content. WT (n = 7), KO (n = 6). *P < .05, **P < .01, ***P < .001, ****P < .0001, *****P = .002.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/24/10.1182_bloodadvances.2018023291/4/m_advances023291f2.png?Expires=1767712394&Signature=yweAUAIV669j76vc4Gl4OZcQw6cWl3C5qxpWQsEQfJRhqJ1j-0HKdmdko-hk4svcQXoEQwUQdKCEeHUi6YHYb6CJBnPne2zcDzijM9Eb838yA6OJdOGZu8F~Dqd0tHB8xaUwjHzlEHRzcKcCl0u9l-8KA0tjF8G1DcDnU-wV6og0TmDUXepmxEIg2m2Bj-oAYFWHzFE4whyWyjdiaD7~gCizatdPhV4lesOJh0Hgq85E1P3S4FLpwfeniOG9zHdN3XwoB2feMq-5-tXhrtHue3Jobqvjpm3SuITY1bboU7of5p0YugVgDo8oKxWIm1ylMvxVn-8DFhKefeknp3YHxQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Secretion is ablated in the absence of SNAP23. (A) Washed platelets at 200 × 109/L were stimulated under nonstirring conditions at 37°C in a Tecan Infinite M200Pro plate reader in the presence of 1/100 Chronolume luciferin luciferase. Median traces of platelets stimulated (arrow) with 0.4 units/mL thrombin (i) and 20 µg/mL collagen-related peptide (CRP) (ii), respectively. (iii) Maximum ATP released upon stimulation (WT [n = 6-8], KO [n = 5]). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (B) Washed platelets at 200 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions at 37°C for 10 minutes before supernatant was harvested, run on a PF4 ELISA, and read at 450 nm. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 3-4), KO (n = 3). (iii) Lysed basal platelets were used to determine total platelet PF4 content (n = 6). (C) Washed platelets at 20 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions for 10 minutes at room temperature in the presence of FITC-conjugated anti–P-selectin. Curves were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 7-8), KO (n = 5). (iii) P-selectin binding at maximal thrombin (Thr; 0.4 units/mL) when costimulated with 10 µM ADP. WT (n = 3-7), KO (n = 4-5). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (D) Washed platelets at 200 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions at 37°C for 10 minutes before supernatant was harvested, run through a β-hexosaminidase colorimetric assay, and read at 405 nm. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 3-4), KO (n = 3). (iii) Lysed basal platelets were used to determine total platelet β-hexosaminidase content. WT (n = 7), KO (n = 6). *P < .05, **P < .01, ***P < .001, ****P < .0001, *****P = .002.
Secretion is ablated in the absence of SNAP23. (A) Washed platelets at 200 × 109/L were stimulated under nonstirring conditions at 37°C in a Tecan Infinite M200Pro plate reader in the presence of 1/100 Chronolume luciferin luciferase. Median traces of platelets stimulated (arrow) with 0.4 units/mL thrombin (i) and 20 µg/mL collagen-related peptide (CRP) (ii), respectively. (iii) Maximum ATP released upon stimulation (WT [n = 6-8], KO [n = 5]). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (B) Washed platelets at 200 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions at 37°C for 10 minutes before supernatant was harvested, run on a PF4 ELISA, and read at 450 nm. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 3-4), KO (n = 3). (iii) Lysed basal platelets were used to determine total platelet PF4 content (n = 6). (C) Washed platelets at 20 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions for 10 minutes at room temperature in the presence of FITC-conjugated anti–P-selectin. Curves were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 7-8), KO (n = 5). (iii) P-selectin binding at maximal thrombin (Thr; 0.4 units/mL) when costimulated with 10 µM ADP. WT (n = 3-7), KO (n = 4-5). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (D) Washed platelets at 200 × 109/L were stimulated with thrombin (i) and CRP (ii) under nonstirring conditions at 37°C for 10 minutes before supernatant was harvested, run through a β-hexosaminidase colorimetric assay, and read at 405 nm. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 3-4), KO (n = 3). (iii) Lysed basal platelets were used to determine total platelet β-hexosaminidase content. WT (n = 7), KO (n = 6). *P < .05, **P < .01, ***P < .001, ****P < .0001, *****P = .002.
The SNAP23 platelet-specific knockout mice unexpectedly showed a macrothrombocytopenia (Table 1), suggesting that absence of SNAP23 may perturb megakaryopoiesis/thrombopoiesis. It was important however to determine in vivo parameters of hemostasis and thrombosis, and we first performed a tail bleed assay to the effect of absence of platelet SNAP23 in hemostasis (Figure 3A). Whereas control mice ceased bleeding within 2 minutes, SNAP23 knockout mice continued to bleed until the experimental end point (10 minutes), indicating a significant role for platelet SNAP23 in hemostasis. We also investigated the loss of SNAP23 on arterial (using a ferric chloride injury model) and venous (using a venous ligation model) thrombosis and found both to be substantially affected (Figure 3B and 3C, respectively). Arterial thrombus formation in the absence of SNAP23 was reduced to a faint monolayer of platelet adhesion at the site of injury (Figure 3Bi) and led to no significant 3-dimensional thrombus formation (Figure 3Bii). Venous thrombus formation was also substantially inhibited (Figure 3Ci-ii), and assessment of wild-type and knockout thrombi by immunohistochemistry showed that SNAP23 ablation also markedly reduced infiltration of CD45+ leukocytes into the remaining small thrombus (Figure 5Ciii), likely as a result of the loss of platelet P-selectin externalization.
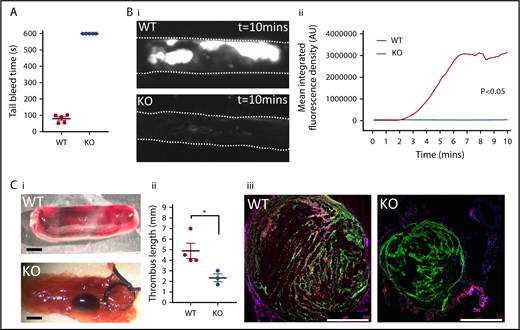
SNAP23 mice show defective hemostasis and arterial and venous thrombosis. (A) Mice anesthetized with a ketamine/xylazine mix had 5 mm of tail tip resected before immediate immersion in 37°C saline. Tail bleeding was followed to the 10-minute experimental end point. N = 5. (B) Mice were infused 0.1 µg/g body weight Dylight-488–conjugated anti-GPIbβ antibody prior to induction of thrombus formation. Thrombus progression was followed for 10 minutes by time-lapse fluorescence microscopy, and representative images of WT (n = 12) and KO (n = 6) of thrombus formation taken under a 4× objective are shown (i) and mean fluorescence intensity for the thrombus is plotted against time after initiation of thrombosis (ii). Data were analyzed by 2-way ANOVA. (C) Mice had their IVCs exposed surgically, and thrombosis was induced by means of a ligature for 48 hours. (i) Representative images are shown of excised vena cavae showing extent of thrombus formation (scale bar = 1 mm). (ii) Thrombus length was recorded after harvest from WT (n = 4) and KO (n = 3) mice. Data were analyzed by Mann-Whitney U test. *P < .05. (iii) Immunohistochemistry was carried out to stain leukocyte marker CD45 (red), platelet marker CD41 (green), and DNA (blue) on venous thrombotic sections, with representative images from WT and KO mice (scale bar = 500 µm).
SNAP23 mice show defective hemostasis and arterial and venous thrombosis. (A) Mice anesthetized with a ketamine/xylazine mix had 5 mm of tail tip resected before immediate immersion in 37°C saline. Tail bleeding was followed to the 10-minute experimental end point. N = 5. (B) Mice were infused 0.1 µg/g body weight Dylight-488–conjugated anti-GPIbβ antibody prior to induction of thrombus formation. Thrombus progression was followed for 10 minutes by time-lapse fluorescence microscopy, and representative images of WT (n = 12) and KO (n = 6) of thrombus formation taken under a 4× objective are shown (i) and mean fluorescence intensity for the thrombus is plotted against time after initiation of thrombosis (ii). Data were analyzed by 2-way ANOVA. (C) Mice had their IVCs exposed surgically, and thrombosis was induced by means of a ligature for 48 hours. (i) Representative images are shown of excised vena cavae showing extent of thrombus formation (scale bar = 1 mm). (ii) Thrombus length was recorded after harvest from WT (n = 4) and KO (n = 3) mice. Data were analyzed by Mann-Whitney U test. *P < .05. (iii) Immunohistochemistry was carried out to stain leukocyte marker CD45 (red), platelet marker CD41 (green), and DNA (blue) on venous thrombotic sections, with representative images from WT and KO mice (scale bar = 500 µm).
Having standardized the platelet count in a functional aggregation assay, we found that the absence of SNAP23 resulted in an attenuated response to maximal concentrations of thrombin and CRP (Figure 4A). Assessment of activation of the αIIbβ3 integrin was then assessed and shown in the knockout to be attenuated in response to thrombin and ablated in response to CRP (Figure 4Bi and ii, respectively). It was important to determine whether this was secondary to loss of δ-granule release and paracrine ADP signaling. Costimulation of SNAP23 knockout platelets with ADP fully rescued the integrin activation defect in response to thrombin; however, for CRP, the rescue was only partial (Figure 4Biii). This suggests that, for thrombin, the defect in integrin activation is solely a product of absence of secreted ADP in the SNAP23 knockout. For CRP, however, the data suggest an additional defect in the GPVI signaling pathway. To understand this difference further, we assessed whether thrombin or CRP-dependent cell calcium responses were altered in the SNAP23 knockout. The response was unchanged upon thrombin stimulation but was reduced in response to CRP (Figure 4C). Likewise, phosphorylation of Syk, a key signal transducer downstream of GPVI, was also reduced in the knockout (Figure 4D). Assessing the surface expression of GPVI, we identified a reduction of ∼27.4% (Table 2), which is likely to explain the signaling defects and the component of the defect in integrin activation that is not rescued by ADP. Interestingly, we also observed a 36.8% increase in platelet α2-integrin expression (Table 2), which might indicate some functional compensation for the reduction in GPVI expression, although this could also be due to the increase in cell surface area because of the macrothrombocytopenia. Platelet αIIb-integrin expression was also slightly reduced (9.6%); although integrin activation in response to dual stimulation with thrombin and ADP yielded comparable values (Figure 4Biii), this is unlikely to be functionally significant. Surface PECAM and GPIb-α levels were not significantly altered (Table 2).
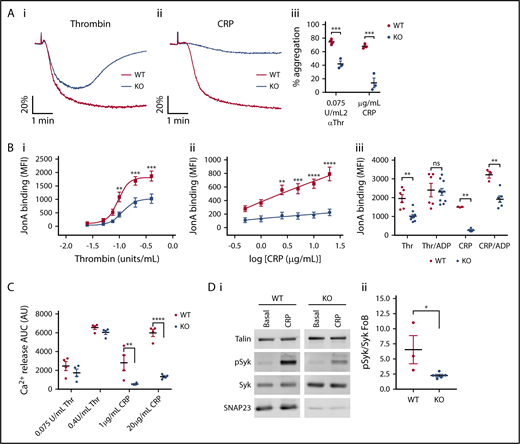
GPVI signaling is affected by the absence of SNAP23. (A) Washed platelets at 200 × 109/L were stimulated with 0.075 units/mL Thr (i) or 2 µg/mL CRP (ii) at 1000 rpm in a Chronolog model 700 aggregometer. (iii) The maximum aggregation within 5 minutes was recorded. Data from at least 3 independent experiments were compared by unpaired Student t test. (B) Washed platelets at 20 × 109/L were stimulated with (i) thrombin and (ii) CRP under nonstirring conditions for 10 minutes at room temperatures in the presence of phycoerythrin-conjugated antibody raised against activated αIIbβ3/CD41 (Jon/A). Curves were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 8), KO (n = 5). (iii) Jon/A binding at maximal thrombin (0.4 units/mL) and maximal CRP (20 µg/mL CRP) were assessed when costimulated with 10 µM ADP. WT (n = 3-6), KO (n = 6-10). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (C) Fura–PE3-labeled platelets at 100 × 109/L were stimulated under nonstirring conditions and analyzed on a Tecan Infinite M200Pro plate reader. At the end of the run, samples were lysed with 0.2% TX100 to give a total value to which values were standardized. n = 4. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (D) Platelets at 400 × 109/L were stimulated for 1 minute with 20 µg/mL CRP under nonstirring conditions at 37°C. Membranes were probed as indicated and imaged on a LI-COR Odyssey CLx. (i) Representative blots of WT (n = 3) and KO (n = 5). (ii) Densitometric analysis of pSyk/Syk ratio as a fold increase over basal. *P < .05, **P < .01, ***P < .001, ****P < .0001. AUC, area under the curve; ns, not significant.
GPVI signaling is affected by the absence of SNAP23. (A) Washed platelets at 200 × 109/L were stimulated with 0.075 units/mL Thr (i) or 2 µg/mL CRP (ii) at 1000 rpm in a Chronolog model 700 aggregometer. (iii) The maximum aggregation within 5 minutes was recorded. Data from at least 3 independent experiments were compared by unpaired Student t test. (B) Washed platelets at 20 × 109/L were stimulated with (i) thrombin and (ii) CRP under nonstirring conditions for 10 minutes at room temperatures in the presence of phycoerythrin-conjugated antibody raised against activated αIIbβ3/CD41 (Jon/A). Curves were compared by 2-way ANOVA with Sidak’s multiple comparisons test. WT (n = 8), KO (n = 5). (iii) Jon/A binding at maximal thrombin (0.4 units/mL) and maximal CRP (20 µg/mL CRP) were assessed when costimulated with 10 µM ADP. WT (n = 3-6), KO (n = 6-10). Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (C) Fura–PE3-labeled platelets at 100 × 109/L were stimulated under nonstirring conditions and analyzed on a Tecan Infinite M200Pro plate reader. At the end of the run, samples were lysed with 0.2% TX100 to give a total value to which values were standardized. n = 4. Data were compared by 2-way ANOVA with Sidak’s multiple comparisons test. (D) Platelets at 400 × 109/L were stimulated for 1 minute with 20 µg/mL CRP under nonstirring conditions at 37°C. Membranes were probed as indicated and imaged on a LI-COR Odyssey CLx. (i) Representative blots of WT (n = 3) and KO (n = 5). (ii) Densitometric analysis of pSyk/Syk ratio as a fold increase over basal. *P < .05, **P < .01, ***P < .001, ****P < .0001. AUC, area under the curve; ns, not significant.
Platelet surface receptors
| Parameter | SNAP23 WT | SNAP23 KO | P | ||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | N | Mean | SEM | N | ||
| αIIb, MFI AU | 6849 | 220.1 | 19 | 6192 | 200.1 | 16 | <.05 |
| α2, MFI AU | 378.9 | 11.6 | 19 | 518.4 | 52.1 | 16 | <.01 |
| GPVI, MFI AU | 641.7 | 30.3 | 19 | 465.9 | 25.1 | 16 | <.001 |
| PECAM, MFI AU | 903.2 | 78.93 | 5 | 1250 | 138.5 | 5 | ns |
| GPIb, MFI AU | 1571 | 564.6 | 5 | 3325 | 650.9 | 5 | ns |
| Parameter | SNAP23 WT | SNAP23 KO | P | ||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | N | Mean | SEM | N | ||
| αIIb, MFI AU | 6849 | 220.1 | 19 | 6192 | 200.1 | 16 | <.05 |
| α2, MFI AU | 378.9 | 11.6 | 19 | 518.4 | 52.1 | 16 | <.01 |
| GPVI, MFI AU | 641.7 | 30.3 | 19 | 465.9 | 25.1 | 16 | <.001 |
| PECAM, MFI AU | 903.2 | 78.93 | 5 | 1250 | 138.5 | 5 | ns |
| GPIb, MFI AU | 1571 | 564.6 | 5 | 3325 | 650.9 | 5 | ns |
P values were determined by unpaired Student t test.
AU, arbitrary units; MFI, median fluorescence intensity.
Because the SNAP23 platelet-specific knockout mouse presented with a macrothrombocytopenia with no granule secretion, it was important to determine whether megakaryopoiesis and platelet production were affected. We determined platelet granule numbers by TEM and found that granule numbers were not significantly altered, nor was there any obvious change in platelet substructure, other than a small increase in size (Figure 5A). With regards to whether megakaryopoiesis is affected in the platelet-specific knockout, the TPO signaling pathway in platelets, as measured by phosphorylated STAT5Y694 (pSTAT5Y694) and phosphorylated JAK (pJAKY1007-1008), was robust and not significantly altered (Figure 5B). Despite this, there was a significant increase in the numbers of megakaryocytes found in the bone marrow of the SNAP23 knockouts (Figure 5C). Staining knockout platelets with pyronin Y to label RNA demonstrated that SNAP23-null platelets have more RNA (Figure 5D). This coupled with their increased size could signify that they are younger (reticulated)18 and that there is a higher turnover of platelets in the SNAP23 platelet-specific knockout, likely to be due to their reduced functionality and an increase in their loss and therefore turnover.
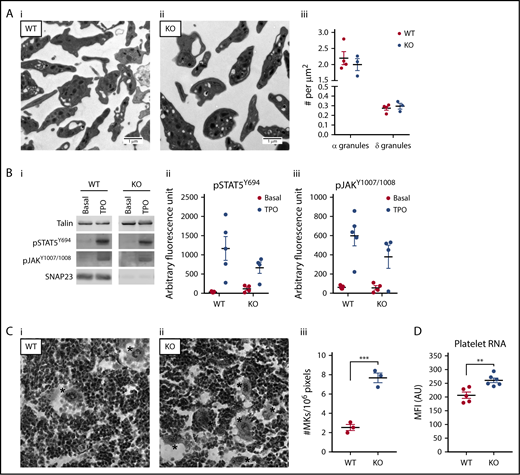
Platelet morphology and thrombopoietin (TPO) signaling are similar, but megakaryopoiesis is increased. (A) TEM sections of representative WT (n = 4) (i) and KO (n = 3) (ii) platelets. (iii) Total platelet section area was measured and α- and δ-granule numbers counted, with granule numbers expressed per unit area of counted section. (B) Platelets at 400 × 109/L were stimulated for 5 minutes with 100 ng/mL TPO under nonstirring conditions at 37°C. Membranes were probed as indicated and imaged on a LI-COR Odyssey CLx. (i) Representative blots of WT (n = 5) and KO (n = 4). Densitometric analysis of pSTAT5Y694 (ii) and pJAKY1007/1008 (iii) expressed as arbitrary fluorescence units as generated by LI-COR Image Studio 5.2. Data were compared by 2-way ANOVA with Sidak’s multiple comparison test. (C) Representative images of WT (i) and KO (ii) hematoxylin and eosin–stained bone marrow imaged using a 40× objective. *Megakaryocytes (MKs). (iii) Megakaryocyte number and bone marrow area were measured using ImageJ 1.46. Megakaryocyte counts were expressed per unit area of counted section. ***P < .001. (D) Washed platelets at 20 × 109/L were labeled with Pyronin Y for 15 minutes at 30°C and immediately analyzed on a BD Accuri C6 Plus. WT (N = 5), KO (N = 6). **P < .01.
Platelet morphology and thrombopoietin (TPO) signaling are similar, but megakaryopoiesis is increased. (A) TEM sections of representative WT (n = 4) (i) and KO (n = 3) (ii) platelets. (iii) Total platelet section area was measured and α- and δ-granule numbers counted, with granule numbers expressed per unit area of counted section. (B) Platelets at 400 × 109/L were stimulated for 5 minutes with 100 ng/mL TPO under nonstirring conditions at 37°C. Membranes were probed as indicated and imaged on a LI-COR Odyssey CLx. (i) Representative blots of WT (n = 5) and KO (n = 4). Densitometric analysis of pSTAT5Y694 (ii) and pJAKY1007/1008 (iii) expressed as arbitrary fluorescence units as generated by LI-COR Image Studio 5.2. Data were compared by 2-way ANOVA with Sidak’s multiple comparison test. (C) Representative images of WT (i) and KO (ii) hematoxylin and eosin–stained bone marrow imaged using a 40× objective. *Megakaryocytes (MKs). (iii) Megakaryocyte number and bone marrow area were measured using ImageJ 1.46. Megakaryocyte counts were expressed per unit area of counted section. ***P < .001. (D) Washed platelets at 20 × 109/L were labeled with Pyronin Y for 15 minutes at 30°C and immediately analyzed on a BD Accuri C6 Plus. WT (N = 5), KO (N = 6). **P < .01.
Discussion
The notion that SNAP23 is a critical regulator of platelet secretion has been regarded as canon in the platelet field since a role for it was first described by Lemons et al and Chen et al in 20009-11 ; however, despite the convincing data, there remained no confirmation in a genetic model, due to early embryonic lethality in the global knockout. Here, we demonstrate for the first time the role of SNAP23 in platelets in a mouse genetic model, which not only validates previous studies but also categorically proves that SNAP23 is the critical regulator for all platelet granule exocytosis. As shown in Figure 2, all platelet granule secretion in response to the stimulation of 2 independent and functionally important receptors (protease-activated receptor and GPVI) by thrombin and CRP is ablated. This includes δ-granule release of ATP, lysosomal release of β-hexosaminidase, and α-granule release of soluble PF4 and surface exposure of the membrane-bound α-granule marker, P-selectin/CD62-P. This is in contrast to other important secretion regulators described for platelets (eg, Syntaxin 114 and Munc18b,5 Munc13-46-8 ) where there was some residual secretion of at least 1 of the granule types, indicating that they could, in some form, be compensated for. There would seem to be no redundancy of function for SNAP23 because all granule secretion is ablated in the knockout, indicating an absolute requirement for SNAP23, and indicating that it is the most critical component of the platelet SNARE complex. Because the SNAP23 knockout platelet granule numbers (Figure 5A) and granule contents (Figures 1 and 2) are not significantly altered, the absence of secretion is not due to a granule biogenesis defect. Also, because the surface exposure of P-selectin in response to thrombin stimulation could not be rescued by costimulation with ADP (to replace lost paracrine P2Y signaling in the knockout platelets8 ; Figure 4Biii), the absence of secretion is unlikely to be due to a loss or reduction in a signaling pathway, and more due to an inability of the granules to fuse with the cell membrane.
It was unexpected to find that the SNAP23 platelet-specific knockout presented with a macrothrombocytopenia (Table 1). Although this may complicate interpretation of in vivo analyses of hemostasis and thrombosis, it was important to determine the effects of platelet-specific SNAP23 gene ablation. We therefore performed tail bleed time analyses and arterial and venous thrombosis assays (Figure 3). The data confirm that mice lacking platelet SNAP23 show a bleeding defect, with no cessation of tail bleeding before the experimental endpoint. Absence of SNAP23 in platelets also ablated arterial thrombosis and reduced venous thrombosis, with reduced leukocyte influx into the venous thrombi. This is likely to be due to a combination of reduced platelet numbers and absent secretion, including the loss of P-selectin exposure and a reduction in the release of localized hemostatic factors, such as fibrinogen and von Willebrand factor. This also indicates that despite the larger size (∼27%) of the SNAP23-null platelets, this does not functionally compensate for the reduced platelet numbers. We have previously demonstrated that in Munc13-4–null mice, which have a normal platelet count and size, but a similar dense granule defect and less pronounced α-granule defect compared with the SNAP23-null platelets, arterial thrombus formation is ablated to a similar extent.7 This suggests that the loss of dense granule secretion in the SNAP23 knockout is likely to be the principal cause of the increased bleeding and reduced arterial thrombus formation, rather than the macrothrombocytopenia. It is also particularly pertinent that there is a substantial reduction in venous thrombosis, because this demonstrates a critical role for platelet secretion, substantially through P-selectin expression possibly, in regulating progression of venous thrombosis.
We explored whether SNAP23-null platelets had functional defects independent of their secretion defects, and as such, platelet aggregation was reduced in response to both thrombin and CRP at maximal agonist concentrations (Figure 4A), as was integrin activation (Figure 4B). ADP rescue experiments for integrin activation showed that the functional defect to thrombin was entirely due to the absence of secreted ADP; however, the functional defect in response to CRP was only partially rescued (Figure 4Biii). Subsequent dissection of signaling events stimulated by thrombin and CRP showed that thrombin elicited robust calcium release in knockout platelets, but CRP did not (Figure 4C). This is likely to be due to the reduction in surface GPVI levels (Table 2) and subsequently reduced phosphorylation of downstream Syk (Figure 4D). It is possible that there are other changes of components of the GPVI signaling pathway because the defect in Syk activation is greater than the ∼27% reduction in GPVI. It could also be that the CRP-specific responses require paracrine signaling from a platelet granule component other than ADP, such as stromal cell–derived factor 1-alpha.19
As described earlier, the SNAP23 platelet-specific knockout mice had an unexpected macrothrombocytopenia (Table 1). Assessment of platelet TPO signaling (Figure 5B) showed no significant defects in the downstream effectors (STAT5 and JAK), and interestingly, bone marrow megakaryocyte numbers were actually increased (Figure 5C), indicating that the thrombocytopenia was not due to reduced megakaryopoiesis. Platelet RNA content has been long used as a readout for “young” or recently generated reticulated platelets18 and SNAP23-null platelets have greater staining for RNA (Figure 5D). Coupled with the increased size associated with “young” platelets, the macrothrombocytopenia observed in the SNAP23 platelet-specific knockout is likely to be due to an increased loss or turnover of platelets, therefore, rather than a defect in their generation.
In summary, this is the first report to demonstrate definitively the function of SNAP23 in platelet activation. SNAP23 is the only gene demonstrated so far for which there is an absolute requirement for secretion from all granule types, and this supports the concept that SNAP23 is a fundamental and central gene in platelet exocytosis. Absence of SNAP23 also revealed a role for the gene in expression or trafficking of important platelet surface receptors (GPVI in particular), and the mice exhibited a macrothrombocytopenia, with increased marrow megakaryocyte numbers and platelet RNA content but normal platelet responsiveness to TPO. This may be indicative of an increase in platelet turnover through bleeding-related loss, presumably because of their severe functional deficit.
Acknowledgments
The authors thank Elizabeth Aitken and David Phillips for general laboratory support and genotyping. They are grateful to the Medical Research Council and the Wolfson Foundation for funding the University of Bristol’s Wolfson Bioimaging Facility.
This work is funded by program and project grant support from the British Heart Foundation (RG/15/16/31758 and PG/15/96/31854).
Authorship
Contribution: C.M.W. and Y.L. designed and performed experiments and wrote the manuscript; E.B. designed and performed experiments; and A.W.P. designed experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alastair W. Poole, School of Physiology, Pharmacology and Neuroscience, University of Bristol, University Walk, Bristol BS8 1TD, United Kingdom; e-mail: a.poole@bristol.ac.uk.

