

Key Points
Gain of 1q and deletions of 17p are associated with perturbations of the TP53 pathway, which contribute to MPN disease progression.
Although gain of 1q is most prevalent in patients with a history of PV, deletion of 17p is most common in patients with a history of PMF.

Abstract
The Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), and the prefibrotic form of primary myelofibrosis (PMF), frequently progress to more overt forms of MF and a type of acute leukemia termed MPN-accelerated phase/blast phase (MPN-AP/BP). Recent evidence indicates that dysregulation of the tumor suppressor tumor protein p53 (TP53) commonly occurs in the MPNs. The proteins MDM2 and MDM4 alter the cellular levels of TP53. We investigated in 1,294 patients whether abnormalities involving chromosomes 1 and 12, which harbor the genes for MDM4 and MDM2, respectively, and chromosome 17, where the gene for TP53 is located, are associated with MPN disease progression. Gain of 1q occurred not only in individuals with MPN-BP but also in patients with PV and ET, who, with further follow-up, eventually evolve to either MF and/or MPN-BP. These gains of 1q were most prevalent in patients with a history of PV and those who possessed the JAK2V617F driver mutation. The gains of 1q were accompanied by increased transcript levels of MDM4. In contrast, 12q chromosomal abnormalities were exclusively detected in patients who presented with MF or MPN-BP, but were not accompanied by further increases in MDM2/MDM4 transcript levels. Furthermore, all patients with a loss of 17p13, which leads to a deletion of TP53, had either MF or MPN-AP/BP. These findings suggest that gain of 1q, as well as deletions of 17p, are associated with perturbations of the TP53 pathway, which contribute to MPN disease progression.
Introduction
The Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), and the prefibrotic form of primary myelofibrosis (PMF), have the potential to progress to more overt forms of MF and a form of acute leukemia (AML) termed MPN-blast phase (MPN-BP). MPN-BP has a dismal prognosis, with a median survival of less than 6 months.1 After 15 years, 6% to 14% of patients with PV and 4% of patients with ET will progress to MF, whereas 7% to 19% of patients with PV and 2% of patients with ET will progress to MPN-BP.2-4 At this time, the mechanisms underlying MPN disease progression remain elusive, but are believed to involve several pivotal factors, including a pro-inflammatory milieu and the acquisition of additional genomic and epigenomic alterations.5-8
One cellular pathway of particular interest in MPN disease progression involves the tumor suppressor tumor protein p53 (TP53), which is located on the short arm of chromosome 17. This gene encodes a DNA-binding protein that responds to DNA damage by either stimulating DNA repair or inducing cell death. Loss or inactivation of TP53 plays a critical role in the pathogenesis of many cancers.9 Although TP53 mutations at low allele burden can be found in chronic MPNs, TP53 loss of heterozygosity is associated with transformation to leukemia, and inactivating mutations of TP53 are found in 20% of patients with MPN-BP.10-14 This is corroborated in the recent publication by Grinfeld et al that presents an approach to classification and personalized prognostication in MPNs and illustrates that those patients with TP53 deletion or mutation are at high risk for transformation to AML and early death.15 In addition, mice that are engineered to possess JAK2V617F and lack TP53 are prone to develop leukemia compared with JAK2V617F mice with wild-type TP53 who develop a picture that resembles either PV or ET.16,17
Two main regulators of TP53 are mouse double-minute homolog 2 (MDM2) and mouse double-minute homolog 4 (MDM4).18 These proteins act in concert to negatively regulate the TP53 pathway by inhibiting TP53 transcription and transactivation, facilitating the export of TP53 from the nucleus and promoting degradation of TP53.19 Downregulation of TP53 activity via overexpression of MDM2 in chronic phase MPNs has been previously reported.20,21
Specific chromosomal abnormalities in MPNs are associated with a poor prognosis, and the longer the duration of the disease, the more likely additional chromosomal abnormalities will be acquired.22,23 At diagnosis, about 25% of patients with PV have abnormal karyotypes, but this increases to 50% at the time of transformation to MF or MPN-BP.24,25 In ET, less than 10% of patients have an abnormal karyotype at diagnosis compared with 15% to 46% at the time of transformation.26
We and others have shown that additional copies of the long arms of chromosome 1 (+1q) are recurrent MPN-associated chromosomal abnormalities, and that the translocation of the whole or part of chromosome 1q to several different recipient chromosomes occurs in MPN-BP.27,28 Andrieux et al23 have reported that 70% of patients with PV-related MF had +1q and attributed the acquisition of +1q to prior treatment with chemotherapy and radioactive phosphorous. Furthermore, Klampfl et al11,29 and Tang et al30 provided additional evidence that +1q abnormalities are associated with MPN-BP and speculated that +1q might lead to increased MDM4 transcripts. Because the long arms of chromosome 12 and chromosome 1 encode the genes for MDM2 and MDM4, respectively, we hypothesized that these abnormalities would lead to altered expression levels of MDM2 and MDM4 transcript. In this study, we have shown that +1q cytogenetic abnormalities occur not only in individuals with MPN-BP but also in patients with PV and ET who with further follow-up eventually evolve to either MF and/or MPN-BP. In fact, we have demonstrated that +1q is the second most common cytogenetic abnormality (6%) after 20q (7%). Furthermore, we have documented that +1q is associated with increased transcripts of MDM4. In addition, rearrangements involving 12 were shown to be primarily associated with MF and were not accompanied by increased transcripts for MDM2, indicating that other gene products are likely involved in the development of this disease phenotype.
Materials and methods
Patients, materials, and methods
PV, ET, MF, and MPN accelerated phase/BP (MPN-AP/BP) were diagnosed by criteria established by the World Health Organization.31 Patients with MPN-AP were defined as having 10% to 19% blasts in their peripheral blood or bone marrow, whereas patients with MPN-BP were defined as having at least 20% blast cells.32
Databases and cytogenetic analysis
Two databases were analyzed: the MPN-Research Consortium (MPN-RC) Trial database and the Tumor Cytogenomics database at the Icahn School of Medicine at Mount Sinai (ISMMS). The MPN-RC database consisted of samples from 373 patients participating in 5 clinical trials that involved patients with high-risk PV, ET, and MF, as well as MPN-AP/BP. The ISMMS database consisted of 921 patients with MPN with cytogenetic analyses. Fourteen patients from the ISMMS database with +1q abnormalities were previously reported.27,28 Bone marrow or unstimulated peripheral blood G-banded metaphase cells were obtained using standard methodology.27 ISCN chromosome nomenclature was used to describe chromosomal abnormalities.33 Interphase FISH was performed as previously described.27 All FISH probes were obtained from Abbott Molecular (Des Plaines, IL). Array comparative genomic hybridization and single-nucleotide polymorphism (CGH+SNP) analysis was performed using GenetiSure Unrestricted −2X400K array (Agilent Technologies, Santa Clara, CA), as previously described.34
MDM2/4 transcript levels
The same peripheral blood and bone marrow mononuclear cells (MNCs) obtained from patients with MF with gains of 1q and 12q rearrangements were used for cytogenetics and determination of MDM2/4 transcript levels. Normal bone marrow MNCs were obtained from AllCells (Alameda, CA). RNA was extracted from MNCs, using RNeasy mini kit (Fisher Scientific, Waltham, MA), and was reverse transcribed using RNA to DNA ecodry kit (Fisher Scientific). qPCR was performed using SYBR green master mix (Fisher Scientific) to test transcript level of human MDM2, MDM4, and PSMB2 (localized at the short arms of #1, at 1p34.3) as the housekeeping gene (Qiagen, Germantown, MD).
Sample processing, sequencing, and mutational analysis
High-throughput sequencing with a targeted deep sequencing assay of 585 genes (HemePACT) was performed on 355 patients participating in the MPN-RC clinical trials, as previously described.35 Driver mutation status and next-generation sequencing (NGS) analyses were performed by the hospital’s molecular diagnostic laboratory or by commercial vendors (Genoptix, INC, and Foundation medicine). NGS became commercially available at Mont Sinai only within the last 5 years; therefore, driver mutations were not available for many patients because of their being studied before this technology was available.
Statistical analysis
Statistical analysis between groups was performed using the Fisher exact test. For RT-PCR studies, statistical significance between transcript levels was determined using the Student t test. Statistical significance was defined as P < .05.
IRB approval
The Institutional Review Board of the Mount Sinai School of Medicine approved this research. It is in compliance with the Declaration of Helsinki.
Results
Gains of the long arms of chromosome 1 (+1q) and structural rearrangements of the long arms of chromosome 12 are recurrent abnormalities in patients with advanced forms of MPN
Between the MPN-RC trial database and the ISMMS database, the karyotypes of a total of 1,294 patients with MPN were reviewed (Table 1). In the MPN-RC database, 25% of the 373 patients were found to have an abnormal karyotype. In the ISMMS database, 36% of the 921 patients were chromosomally abnormal. The most frequent chromosomal abnormality among the 1,294 patients was del(20)(q11q13), which occurred in 7% of total patients and was present alone or in combination with other abnormalities. Surprisingly, +1q/dup(1q) was the second most frequent cytogenetic abnormality, being observed in 6% of total patients. In contrast, 12q rearrangements and deletion of 17p13 each occurred in 2% of total patients. Other frequent recurrent abnormalities included interstitial deletion of the long arm of chromosome 13 (2%), +8 sole (2%), +9 sole (2%), and simultaneously occurring +8 and +9 (1%).
Prevalence of +1q, 12q rearrangements and deletions of 17p within the MPN-RC Trials database and the ISMMS database and the association of +1q with progressive disease
| MPN-RC database | ISMMS database | Total | |
|---|---|---|---|
| No. of patients | 373 | 921 | 1294 |
| Chromosomally abnormal, n (%) | 93 (25) | 330 (36) | 423 (33) |
| +1q, n (%) | 15 (4) | 57 (6) | 72 (6) |
| 12q rearrangements, n (%) | 7 (2) | 17 (2) | 24 (2) |
| del(17)(p13), n (%) | 9 (2) | 21 (2) | 30 (2) |
| MPN-RC database | ISMMS database | Total | |
|---|---|---|---|
| No. of patients | 373 | 921 | 1294 |
| Chromosomally abnormal, n (%) | 93 (25) | 330 (36) | 423 (33) |
| +1q, n (%) | 15 (4) | 57 (6) | 72 (6) |
| 12q rearrangements, n (%) | 7 (2) | 17 (2) | 24 (2) |
| del(17)(p13), n (%) | 9 (2) | 21 (2) | 30 (2) |
Gain of 1q was attributed to 3 different events: as an unbalanced translocation with the breakpoint occurring at 1q12∼21, a duplication of the 1q region inserted within 1q, and/or a jumping +1q to different partner chromosomes (Figure 1A-C,H). In several patients with the whole-arm 1q translocation array, CGH+SNP analysis demonstrated that patients with +1q had a gain of 105 MB between 1q21 and 1q44 regions. Duplication of the 1q21-q31/q41 region was found in 6 patients (Figure 1A,I). Because MDM4 is localized to 1q32.1, we examined cytogenetically the most minimally gained 1q region among the patients with dup(1q) and jumping +1q translocations. This region was identified between 1q21 and q32.
![Figure 1. Structural chromosomal abnormalities involving chromosomes 1, 12, and 17 in patients with MPN and progression of disease. (A-G) Partial karyotypes showing gains of 1q and 12q structural rearrangements. The most frequent unbalanced translocations identified were +der(9)t(1;9)(q21;q12), resulting in 3 copies of 1q and 3 copies of 9p. Extra copies of 1q were also observed as the whole-arm or part of the +1q participated in unbalanced translocations to chromosomes 1, 13, 14, 15, 18, 19, 21, or 22 (26 patients). (A) Two different 1q duplications were inserted in dup(1q) in a patient with MF. The first region involved 1q12-q32, and the second region involved 1q21-32, showing both duplication of q12 (red) and q21 (aqua). (B) A partial karyotype from a patient with PV showing the most frequent gain of 1q, der(9)t(1;9)(q21;q12), resulting in 3 copies of 9p and JAK2. (C) A partial karyotype showing isochromosome 1q in a patient with MF, resulting in 4 copies of 1q and MDM4. (D) The most frequent 12q abnormalities are inversions of chromosome 12. In this partial karyotype, as a result of pericentric inversion, ETV6 (yellow), normally residing on the short arms (12p13.2), has been relocated to 12q15, where MDM2 is localized. (E) The second most common 12q rearrangements were balanced translocations between 12q and the following partner chromosomes: 2, 5, 8, and 10. A partial karyotype showing a balanced t(12;17)(q15;q21) translocation involving the breakpoint at 12q15, as does the t(10;12) (see panel F) observed in 3 patients with MF, is shown here. (F) A partial karyotype showing a balanced translocation t(10;12)(q11.2;q15) in a patient with MF. Two patients with MF were identified with the identical translocation. (G) A different type of t(12;17) translocation identified in a patient who progressed to MPN-AP/BP. The patient had a complex karyotype, and this partial karyotype shows both the 12q15 and the 17p13, where TP53 resides, were rearranged in the same clone. (H) Sequential cytogenetic studies spanning 16 years. Initial cytogenetic study (1996) at the time of PV diagnosis revealed a gain of isochromosome 9p, resulting in tetrasomy 9p (and 4 copies of JAK2) in 97% of metaphase cells. An additional 20 cytogenetic studies revealed gain of 1q [der(6)t(1;6)] initially in 10% of i(9p) cells for the first time 7 years after the diagnosis. As the number of cells harboring +1q increased, the patient progressed to MF, and finally to MPN-BP. This progression was also accompanied by jumping 1q, whereby long arms of chromosome 1 were translocated to 4 different recipient chromosomes, leading to the 6 copies of 1q and transformation of the disease to MPN-BP. (I) At the time of ET diagnosis, the patient had 36% of cells with dup(1q). Within 2 years, the patient progressed to PV, which was accompanied by 50% of interphase cells showing gain of CKS1B (aqua) at 1q21 and 3 copies of ABL2 (green) at 1q25, as detected by FISH. The patient progressed to MF 5 years after the diagnosis of ET, at which time 95% of the cells harbored duplication of 1q21-q31 region.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/24/10.1182_bloodadvances.2018024018/4/m_advances024018f1.png?Expires=1769079694&Signature=druczAd53InRSrBivWyicfTyJX3sJDP2n7PLeenwg3O6OekcK7u74GGm~fmh6SGbCDvJ4hfZGMqIe-lr~jNUbcKkwDEzAArf3oKLrri6wJ9DOugO1G1~8FKdX4EYL0~obUhnto9Z5oOUtltdgTxsfR59jYZoyHcdaJERxVrWV6Hk5L2JDEA9odo4HlZMVBUlVm4tQB3NvtFT8M6SqffJNJZB7d2E4aIgDqEMoK7ojHj9tHCNC5rtVc53MfVq~1PgqZ7WJ-VHJEorysxgSR3c9ey2XXj47BPB7WM1UeTzZ-jNOMQvVgqfs6C47UxWCBUk08~2xKDuksp3yU~2Ovzvew__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structural chromosomal abnormalities involving chromosomes 1, 12, and 17 in patients with MPN and progression of disease. (A-G) Partial karyotypes showing gains of 1q and 12q structural rearrangements. The most frequent unbalanced translocations identified were +der(9)t(1;9)(q21;q12), resulting in 3 copies of 1q and 3 copies of 9p. Extra copies of 1q were also observed as the whole-arm or part of the +1q participated in unbalanced translocations to chromosomes 1, 13, 14, 15, 18, 19, 21, or 22 (26 patients). (A) Two different 1q duplications were inserted in dup(1q) in a patient with MF. The first region involved 1q12-q32, and the second region involved 1q21-32, showing both duplication of q12 (red) and q21 (aqua). (B) A partial karyotype from a patient with PV showing the most frequent gain of 1q, der(9)t(1;9)(q21;q12), resulting in 3 copies of 9p and JAK2. (C) A partial karyotype showing isochromosome 1q in a patient with MF, resulting in 4 copies of 1q and MDM4. (D) The most frequent 12q abnormalities are inversions of chromosome 12. In this partial karyotype, as a result of pericentric inversion, ETV6 (yellow), normally residing on the short arms (12p13.2), has been relocated to 12q15, where MDM2 is localized. (E) The second most common 12q rearrangements were balanced translocations between 12q and the following partner chromosomes: 2, 5, 8, and 10. A partial karyotype showing a balanced t(12;17)(q15;q21) translocation involving the breakpoint at 12q15, as does the t(10;12) (see panel F) observed in 3 patients with MF, is shown here. (F) A partial karyotype showing a balanced translocation t(10;12)(q11.2;q15) in a patient with MF. Two patients with MF were identified with the identical translocation. (G) A different type of t(12;17) translocation identified in a patient who progressed to MPN-AP/BP. The patient had a complex karyotype, and this partial karyotype shows both the 12q15 and the 17p13, where TP53 resides, were rearranged in the same clone. (H) Sequential cytogenetic studies spanning 16 years. Initial cytogenetic study (1996) at the time of PV diagnosis revealed a gain of isochromosome 9p, resulting in tetrasomy 9p (and 4 copies of JAK2) in 97% of metaphase cells. An additional 20 cytogenetic studies revealed gain of 1q [der(6)t(1;6)] initially in 10% of i(9p) cells for the first time 7 years after the diagnosis. As the number of cells harboring +1q increased, the patient progressed to MF, and finally to MPN-BP. This progression was also accompanied by jumping 1q, whereby long arms of chromosome 1 were translocated to 4 different recipient chromosomes, leading to the 6 copies of 1q and transformation of the disease to MPN-BP. (I) At the time of ET diagnosis, the patient had 36% of cells with dup(1q). Within 2 years, the patient progressed to PV, which was accompanied by 50% of interphase cells showing gain of CKS1B (aqua) at 1q21 and 3 copies of ABL2 (green) at 1q25, as detected by FISH. The patient progressed to MF 5 years after the diagnosis of ET, at which time 95% of the cells harbored duplication of 1q21-q31 region.
Structural chromosomal abnormalities involving chromosomes 1, 12, and 17 in patients with MPN and progression of disease. (A-G) Partial karyotypes showing gains of 1q and 12q structural rearrangements. The most frequent unbalanced translocations identified were +der(9)t(1;9)(q21;q12), resulting in 3 copies of 1q and 3 copies of 9p. Extra copies of 1q were also observed as the whole-arm or part of the +1q participated in unbalanced translocations to chromosomes 1, 13, 14, 15, 18, 19, 21, or 22 (26 patients). (A) Two different 1q duplications were inserted in dup(1q) in a patient with MF. The first region involved 1q12-q32, and the second region involved 1q21-32, showing both duplication of q12 (red) and q21 (aqua). (B) A partial karyotype from a patient with PV showing the most frequent gain of 1q, der(9)t(1;9)(q21;q12), resulting in 3 copies of 9p and JAK2. (C) A partial karyotype showing isochromosome 1q in a patient with MF, resulting in 4 copies of 1q and MDM4. (D) The most frequent 12q abnormalities are inversions of chromosome 12. In this partial karyotype, as a result of pericentric inversion, ETV6 (yellow), normally residing on the short arms (12p13.2), has been relocated to 12q15, where MDM2 is localized. (E) The second most common 12q rearrangements were balanced translocations between 12q and the following partner chromosomes: 2, 5, 8, and 10. A partial karyotype showing a balanced t(12;17)(q15;q21) translocation involving the breakpoint at 12q15, as does the t(10;12) (see panel F) observed in 3 patients with MF, is shown here. (F) A partial karyotype showing a balanced translocation t(10;12)(q11.2;q15) in a patient with MF. Two patients with MF were identified with the identical translocation. (G) A different type of t(12;17) translocation identified in a patient who progressed to MPN-AP/BP. The patient had a complex karyotype, and this partial karyotype shows both the 12q15 and the 17p13, where TP53 resides, were rearranged in the same clone. (H) Sequential cytogenetic studies spanning 16 years. Initial cytogenetic study (1996) at the time of PV diagnosis revealed a gain of isochromosome 9p, resulting in tetrasomy 9p (and 4 copies of JAK2) in 97% of metaphase cells. An additional 20 cytogenetic studies revealed gain of 1q [der(6)t(1;6)] initially in 10% of i(9p) cells for the first time 7 years after the diagnosis. As the number of cells harboring +1q increased, the patient progressed to MF, and finally to MPN-BP. This progression was also accompanied by jumping 1q, whereby long arms of chromosome 1 were translocated to 4 different recipient chromosomes, leading to the 6 copies of 1q and transformation of the disease to MPN-BP. (I) At the time of ET diagnosis, the patient had 36% of cells with dup(1q). Within 2 years, the patient progressed to PV, which was accompanied by 50% of interphase cells showing gain of CKS1B (aqua) at 1q21 and 3 copies of ABL2 (green) at 1q25, as detected by FISH. The patient progressed to MF 5 years after the diagnosis of ET, at which time 95% of the cells harbored duplication of 1q21-q31 region.
Types of 12q rearrangements are summarized in Figure 1D-G. The 12q15 breakpoint region where MDM2 is located was involved in 12/24 patients with 12q rearrangements. The other 50% of patients had the breakpoints at 12q13 (25%) or q21-24 (25%). Gain of 1q/dup(1q) and rearrangements with 12q were mutually exclusive, with the exception of 1 patient with an unbalanced translocation der(12)t(1;12)(q25;q13).
Gain of 1q is associated with MPN disease progression
The early stages of MPNs (PV and ET) are characterized by hyperproliferation and effective blood cell production, leading to erythrocytosis, thrombocytosis, and leukocytosis. These disorders are thought to represent the proliferative phase of the MPNs. As shown in Table 2, +1q was more prevalent in more advanced forms of MPNs (MF and MPN-AP/BP) than PV and ET (P < .001). Two percent of patients with PV and ET had +1q, whereas 9% of patients with MF (PMF or PV/ET-related MF) and 43% of MPN-AP/BP had +1q.
Prevalence of +1q in different stages of MPN
| MPN-RC database | ISMMS database | Total | |
|---|---|---|---|
| Proliferative phases of MPNs (PV, ET), n | 231 | 550 | 781 |
| No. (%) with +1q | 3 (1) | 10 (2) | 13 (2) |
| Myelofibrosis, n | 113 | 343 | 456 |
| No. (%) with +1q | 10 (9) | 33 (10) | 43 (9)* |
| MPN-AP/BP, n | 9 | 28 | 37 |
| No. (%) with +1q | 2 (22) | 14 (50) | 16 (43)† |
| MPN-RC database | ISMMS database | Total | |
|---|---|---|---|
| Proliferative phases of MPNs (PV, ET), n | 231 | 550 | 781 |
| No. (%) with +1q | 3 (1) | 10 (2) | 13 (2) |
| Myelofibrosis, n | 113 | 343 | 456 |
| No. (%) with +1q | 10 (9) | 33 (10) | 43 (9)* |
| MPN-AP/BP, n | 9 | 28 | 37 |
| No. (%) with +1q | 2 (22) | 14 (50) | 16 (43)† |
Incidence of gain of 1q in patients with MF vs PV and ET, P < .0001.
Incidence of gain of 1q in patients with MPN-AP/BP vs PV and ET, P < .0001.
More than 50% of patients who acquired +1q had an initial diagnosis of PV (Table 3; PV vs ET, P < .001; PV vs PMF, P = .002). In most of these patients with a history of PV, the +1q abnormality was first observed at the time they had progressed to MF or MPN-AP/BP (Table 3). Gain of 1q was less frequently observed in patients with ET and PMF (Tables 3 and 4).
Initial MPN diagnosis of patients who acquired +1q,12q rearrangements and del(17p)
| Chromosomal abnormality, n (%) | PV | ET | PMF |
|---|---|---|---|
| +1q | 39/72 (54)* | 13/72 (18) | 20/72 (28) |
| 12q rearrangement | 2/24 (8) | 1/24 (4) | 21/24 (88)† |
| del(17)(p13) | 8/30 (27) | 5/30 (17) | 17/30 (57)‡ |
| Chromosomal abnormality, n (%) | PV | ET | PMF |
|---|---|---|---|
| +1q | 39/72 (54)* | 13/72 (18) | 20/72 (28) |
| 12q rearrangement | 2/24 (8) | 1/24 (4) | 21/24 (88)† |
| del(17)(p13) | 8/30 (27) | 5/30 (17) | 17/30 (57)‡ |
Incidence of patients who acquired gain of 1q with prior history of PV vs ET, P < .001; PV vs PMF, P = .002.
Incidence of patients who acquired 12q rearrangement with prior history of PMF vs PV, P < .0001; PMF vs ET, P < .0001.
Incidence of patients who acquired del(17)(p13) with prior history of PMF vs PV, P = .02; PMF vs ET, P = .001.
Stage of MPNs at which +1q, 12q rearrangements and del(17p) were first observed
| Chromosomal abnormality, n (%) | No. of patients | PV | ET | PMF | PV-related MF | ET-related MF | MPN-AP/BP |
|---|---|---|---|---|---|---|---|
| +1q | 72 | 12 (17) | 2 (3) | 18 (25) | 17 (24) | 7 (10) | 16 (22) |
| 12q rearrangement | 24 | 0 (0) | 0 (0) | 18 (75) | 1 (4) | 1 (4) | 4 (17) |
| del(17)(p13) | 30 | 0 (0) | 0 (0) | 15 (50) | 3 (10) | 2 (7) | 10 (33) |
| Chromosomal abnormality, n (%) | No. of patients | PV | ET | PMF | PV-related MF | ET-related MF | MPN-AP/BP |
|---|---|---|---|---|---|---|---|
| +1q | 72 | 12 (17) | 2 (3) | 18 (25) | 17 (24) | 7 (10) | 16 (22) |
| 12q rearrangement | 24 | 0 (0) | 0 (0) | 18 (75) | 1 (4) | 1 (4) | 4 (17) |
| del(17)(p13) | 30 | 0 (0) | 0 (0) | 15 (50) | 3 (10) | 2 (7) | 10 (33) |
Partial or full trisomy 1q as a sole abnormality was identified in 46% (33/72) of patients. In the remaining patients (39/72; 54%), +1q was associated with additional chromosomal abnormalities, but a clear association between +1q and another chromosomal abnormality was not apparent.
Sequential cytogenetic studies (more than 3) were available in 14 patients. Figure 1H-I illustrate chronological acquisition of +1q as the MPN progressed. One patient (Figure 1H) was diagnosed with PV with an iso-chromosome 9p that appeared in 97% of cells. The initial acquisition of +1q in 10% of cells was observed 7 years later in the iso(9p) clone. Over time, greater numbers of cells had +1q (10%-100%), including +1q jumping to 4 different recipient chromosomes (1, 6, 7, and Y chromosome). The patient transformed to MPN-BP 9 years after the initial detection of +1q. In addition, a patient with ET (Figure 1I) had duplication of 1q present in 36% of cells as the sole cytogenetic abnormality. As the disease progressed to PV, the percentages of hematopoietic cells with dup(1q) increased to 50%. After 3 years, the patient developed MF, and 95% of the cells harbored dup(1q). These studies demonstrate that the burden of cells with +1q gradually increases over time in patient with MPN and frequently precedes evolution to a more advanced form of MPN.
Rearrangements of 12q are associated with PMF
As shown in Tables 3 and 4, rearrangements of 12q were primarily associated with PMF. Seventy-five percent of the 24 patients with 12q rearrangements (Table 4) had PMF, whereas only 1 patient had PV-related MF and 1 patient had ET-related MF. Four patients had MPN-AP/BP, 3 of which had progressed from PMF. Structural 12q abnormalities included primarily either balanced translocations or inversions within chromosome 12, which initially appeared in 90% to 100% of the hematopoietic cells. Moreover, in 84% of the patients, the 12q abnormality was the sole abnormality; only 4 patients had additional karyotypic changes in the clone with 12q. Three patients with MF had recurrent t(12;17) (q15;17variable), with the same breakpoint at 12q15 and variable breakpoints on chromosome 17 causing loss of 17p13 in 2 patients.
One patient with a 12q abnormality had serial studies. At the diagnosis of PV, the patient and had an iso-chromosome involving the short arm of chromosome 9; i(9)(p10), as well as an iso-chromosome of the long arm of chromosome 9, i(9)(q10), in all 20 metaphase cells. After 29 years, the patient’s disease progressed to MF and 100% of 20 metaphase cells had i(9p); i(9q), a subclone harboring inversion of chromosome 12, was identified in 15% of cells [46,XY,i(9)(p10,i(9)(q10),inv(12)(q14q24.3)]. These studies confirm the association between 12q abnormalities and the evolution to MF.
JAK2617F mutation is associated with +1q
The MPN driver mutations were evaluated for all the patients in the MPN-RC database, but only a subset of patients in the ISMMS database, because many were studied before discovery of these mutations. JAK2V617F was observed in 84% (37/44) of patients with MPN with +1q compared with 23% (3/13) patients with MPN with 12q rearrangements (P < .001). The presence of CALR or MPL mutations was very infrequent in patients with +1q.
Deletions and mutations of TP53
A total of 30 patients (2% of all patients) had a deletion of 17p13, identified either cytogenetically or by interphase FISH (Table 1). All these patients had either MF or MPN-AP/BP (Table 4). As shown in Table 3, the majority of TP53 deletions were present in patients who had a diagnosis of PMF (PMF vs PV P = .02; PMF vs ET P = .001).
The most frequent loss of 17p13 was caused by the presence of monosomy 17, followed by deletion of 17p13, formation of isochromosome 17q and therefore loss of 17p, and different balanced or unbalanced translocations involving the 17p13 region. In 97% (29/30) of patients, these 17p structural abnormalities involved 1 chromosome 17 (heterozygous). Only 1 patient had homozygous loss of 17p13. One patient had a normal karyotype but a crypticTP53 deletion detected by FISH. Loss of 17p13 was identified as a sole abnormality in only 17% (5/30) of patients. Nine patients with TP53 deletions had NGS data, and 1 of these patients had a TP53 mutation.
NGS was performed on 355 patients enrolled in 5 MPN-RC clinical trials. Eight (2%) of these patients had TP53 mutations. The mean allelic burden for the 6 patients was 27% (PV: 6%, 33%, 37%, and 48%; ET: 6%; MF, 32%). The mean allelic burden in the 2 patients with MPN-AP/BP was 43% (5% and 81%, respectively). Overall, our cohort of patients with TP53 mutations was too small to determine whether TP53 mutations are associated with specific cytogenetic abnormalities; however, 2 of these 8 patients were cytogenetically abnormal and 6 had a normal karyotype.
Association of +1q and 12q abnormalities with deletion of TP53 and TP53 mutations
Co-occurrence of a TP53 deletion with +1q was observed in 4 patients: 3 patients had PMF and 1 patient had ET that progressed to MF and subsequently progressed to MPN-AP/BP. The loss of TP53 in 2 of these patients was the result of jumping +1q as a consequence of der(17)(t(1;17)(q21;p11), whereby the long arms of chromosome 1 were translocated just above the centromere of chromosome 17 with a loss of the entire short arms of chromosome 17. There were 3 patients with 12q rearrangements and TP53 deletions: 1 had PMF and the other progressed to MPN-AP/BP after MF.
In the MPN-RC clinical trials, none of the 8 patients with TP53 mutations had +1q or 12q abnormalities. In the ISMMS cohort, 13 patients with gain of 1q had NGS data, and 1 of these patients was found to have a TP53 mutation showing that +1q and TP53 mutations are infrequent but not mutually exclusive. One patient with a 12q abnormality had an NGS panel, and a TP53 mutation was not present.
Upregulation of MDM4 transcript levels in patients with +1q
MDM4 transcript levels were measured in the MNCs of patients with MPN with +1q, 12q abnormalities and healthy control patients (Figure 2A). There was a 1.64-fold increase in MDM4 transcript in patients with MPN with gain of 1q (n = 9) compared with healthy individuals (n = 10; P = .004), but not in patients with a 12q rearrangement (n = 5). A significant increase in the transcript levels of MDM2 in patients with MPN with either +1q (n = 9) or 12q rearrangements (n = 5) compared with normal health donors (n = 5) was not observed (Figure 2B).
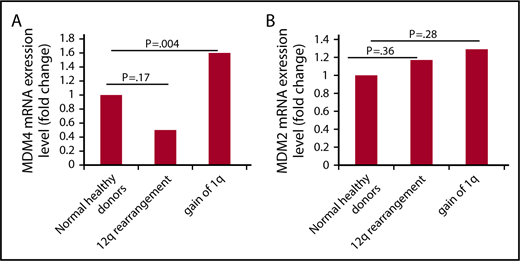
MDM4/MDM2 transcript levels in patients with MPN patients with +1q or 12q rearrangements. Gain of 1q in patients with MF is associated with increase in MDM4 transcript level. MDM4 and MDM2 transcript levels were measured in the mononuclear cells of patients with MPN with a gain of 1q, patients with MPN with a 12q rearrangement, and healthy donors. (A) There was a 1.64-fold increase in MDM4 transcript in patients with MPN, with gain of 1q (n = 9) as compared with healthy individuals (n = 10; P=.004), but not in patients with 12q rearrangements (n = 5). (B) There was no increase in MDM2 transcript in patients with MPN with 12q rearrangement (n = 5) or gain of 1q (n = 9) compared with healthy individuals (n = 5). MDM2, Mouse Double Minute homolog 2; MDM4, Mouse Double Minute homolog 4.
MDM4/MDM2 transcript levels in patients with MPN patients with +1q or 12q rearrangements. Gain of 1q in patients with MF is associated with increase in MDM4 transcript level. MDM4 and MDM2 transcript levels were measured in the mononuclear cells of patients with MPN with a gain of 1q, patients with MPN with a 12q rearrangement, and healthy donors. (A) There was a 1.64-fold increase in MDM4 transcript in patients with MPN, with gain of 1q (n = 9) as compared with healthy individuals (n = 10; P=.004), but not in patients with 12q rearrangements (n = 5). (B) There was no increase in MDM2 transcript in patients with MPN with 12q rearrangement (n = 5) or gain of 1q (n = 9) compared with healthy individuals (n = 5). MDM2, Mouse Double Minute homolog 2; MDM4, Mouse Double Minute homolog 4.
Discussion
The current study provides data that support the hypothesis that events that lead to dysfunction of the TP53 pathway are associated with disease progression in patients with MPN. These events include deletion of TP53, mutations of TP53, and upregulation of MDM2/4, which downregulate wild-type TP53 activity. Prior reports have suggested that the presence of additional copies of 1q are associated with advanced forms of MPNs.28,29,36 In this report, we focus on patients with deletions of TP53 and +1q abnormalities, as MDM4 is present on this portion of chromosome 1. Although 54% of patients in this report had +1q associated with additional chromosomal aberrations, in 46%, +1q represented the sole abnormality. We observed that increasing numbers of cells with +1q over time was associated with disease progression. This is best illustrated by the longitudinal study of a patient with PV who, over the course of 16 years, had a progressive increase in the number of cells with +1q until the number of cells harboring +1q reached 100% and subsequently progressed to MPN-BP.
The MIPPS70 risk scoring system, which was designed to predict the prognosis of patients with MF, concluded that the presence of +1q as a sole abnormality was associated with a favorable prognosis.37 Our observations are partially in conflict with these conclusions. We demonstrated that the presence of +1q at a single point in time has limited use as a prognostic parameter, but that increases in the numbers of hematopoietic cells with this abnormality over time frequently accompanies MPN disease progression.
The association of +1q and disease progression is not unique to the MPNs; numerous publications have reported that +1q is a secondary cytogenetic abnormality and has a negative effect on prognosis in patients with multiple myeloma,38-40 as well as individuals with myelodysplastic disorders,41 and less frequently in AML.42 In myeloma, as in the MPNs, the primary mechanism of +1q is the translocation of the long arm to single or multiple chromosomes. Another mechanism of jumping +1q21 translocation in multiple myeloma involves the focal amplification of 1q21, but this event has not been observed (unpublished observation) in patients with MPN.43
We have previously shown that MDM2 is upregulated in CD34+ cells of both PV and MF patients.20 JAK2V617F negatively regulates TP53 in MPNs by increasing the expression of La antigen, which increase the translation of MDM2.21 We showed here that +1q was associated with increased transcripts of MDM4, likely leading to further downregulation of TP53 activity. Elevated MDM4 levels have been previously found in other hematologic malignancies, including de novo AML.44 There are numerous other oncogenes that are present on 1q, which could also contribute to disease progression. The overexpression of CKS1B, involved in cell cycle regulation, located at band 1q21 has been proposed as the culprit responsible for disease progression in patients with myeloma with +1q.45,46 Although increased CKS1B or another yet-to-be implicated gene present on 1q might also contribute to MPN disease progression; we favor the hypothesis that this abnormality leads to further dysregulation of the wild-typeTP53 pathway by increasing MDM4 activity. In support of this hypothesis is the observation that MF and acute leukemia develop in JAK2V617F transgenic mice, which lack TP53 compared with JAK2V617F mice with wild-type TP53.16
The +1q abnormality in patients with MPN was closely associated with the presence of JAK2V617F. Vainchenker and colleagues have previously reported that JAK2V617F leads to increased genomic instability, which could contribute to the outcomes of patients with this +1q abnormality.47,48 We found that that 24% of our patients with +1q (17/72) had der(9)t(1;9)(q21;q12), resulting in 3 copies of 1q and 3 copies of JAK2, suggesting chromosomal instability. The exact mechanism underlying this close association requires further investigation.
Chromosome 12 abnormalities have been previously reported to occur in patients with MPN.49 One prior report indicated that chromosome 12q rearrangements were found primarily in PV-related MF,50 whereas others reported that 2 specific 12q rearrangements at 12q15 and 12q24 were associated with PMF.51,52 Here, the 12q rearrangements were found most commonly in patients with PMF, but were also observed in 1 patient with ET at the time of evolution to ET-related MF. The 12q rearrangements occurred in 84% of these patients as a single abnormality and were associated in 13% of patients with progression to MPN-BP. These 12q abnormalities did not result in increased copy numbers of MDM2, but were associated in 3 patients with deletion of TP53. Furthermore, 12q abnormalities were present most frequently in the majority of hematopoietic cells at diagnosis. The molecular consequences of 12q translocations require further study. Earlier studies have reported that 12q15 structural abnormalities in MF are associated with increased transcripts of the high mobility AT-hook 2 (HMGA2) gene.53 These investigators, however, indicated that HMGA2 transcripts were increased in patients with or without 12q15 chromosomal abnormalities, leading us to question whether 12q structural abnormalities were responsible for the increased transcript levels. This is further supported by more recent studies demonstrating increased HMGA2 expression in patients with PMF lacking 12q abnormalities.54,55
Thirty patients (2% of total patients) in this report had deletions of 17p13, which leads to loss of TP53, whereas 8 (2%) had inactivating mutations of TP53. Of the 30 patients with deletions of 17p13, 7 also had either +1q (4 patients) or 12q abnormalities (3 patients), and 1 patient had a TP53 mutation. One patient with +1q was also found to have a TP53 mutation. The TP53 mutations were observed in patients with PV, MF, and MPN-AP/BP whereas the deletions of TP53 were associated exclusively with MF and MPN-AP/BP. These findings indicate that a variety of events leading to loss of TP53 function can occur individually or in combination in patients with MPN as their disease evolves.
Cytogenetic analysis and NGS are complementary methods to detect genetic abnormalities in patients with MPN. This study supports the use of cytogenetic analysis for risk stratification in MPNs. Gain of 1q, 12q translocations/rearrangements and del17p in MPNs should be considered poor risk factors and should be added to risk stratification systems to help in prognosticating for likelihood of disease progression and consideration for hematopoietic stem cell transplantation.
In summary, we and others have provided evidence that a series of genetic events occur in patients with MPN, which result in decreased activity of the TP53 pathway and likely contribute to MPN disease progression (Figure 3). These events include deletion of TP53, mutations of TP53 and upregulation of the negative regulators of TP53, MDM2, and MDM4.20,56 On the basis of these findings, we speculate that drugs that antagonize MDM2/4 and upregulate wild-type TP53 might be useful not only in treating the underlying MPN but also in preventing disease progression in patients with wild-type TP53. In support of this hypothesis are the results of a recent a phase 1 trial using the MDM2 inhibitor idasanutlin in patients with PV, showing an unusually high degree of clinical activity.57 Furthermore, the outcomes of patients with +1q and increased MDM4 transcripts might specifically be improved by use of a stapled protein that inhibits MDM2 as well as MDM4.58,59

Proposed role of TP53 dysregulation in the evolution of the MPNs. Decreased TP53 activity can result from several genomic alterations in the MPN clone and is hypothesized to be a crucial driver of MPN disease progression. Mutations and deletions (del17p) of the TP53 gene can directly result in decreased TP53 activity. MDM2 and MDM4 are negative regulators of TP53 that decrease transcription and increase degradation of TP53. Upregulation of these regulators ultimately decreases p53 activity. In MPNs, there are increased MDM2 protein levels resulting from increased MDM2 translation stimulated by JAK2V617 through the La autoantigen. In addition, gain of 1q results in upregulation of MDM4 transcript levels.
Proposed role of TP53 dysregulation in the evolution of the MPNs. Decreased TP53 activity can result from several genomic alterations in the MPN clone and is hypothesized to be a crucial driver of MPN disease progression. Mutations and deletions (del17p) of the TP53 gene can directly result in decreased TP53 activity. MDM2 and MDM4 are negative regulators of TP53 that decrease transcription and increase degradation of TP53. Upregulation of these regulators ultimately decreases p53 activity. In MPNs, there are increased MDM2 protein levels resulting from increased MDM2 translation stimulated by JAK2V617 through the La autoantigen. In addition, gain of 1q results in upregulation of MDM4 transcript levels.
Acknowledgments
This work was supported by the National Institutes of Health, National Cancer Institute (MPN Research Consortium Grant 2P01CA108671-10A1), Cancer Center Support Grant/Core Grant (P30 CA008748) to Memorial Sloan Kettering Cancer Center, and National Cancer Institute Grant 1K08CA188529-01(R.K.R.), and the Leukemia Lymphoma Society (LLS Translational Research Program R6506-17).
Authorship
Contribution: B.K.M., R.H., and V.N. designed the study, analyzed the data, and prepared the manuscript; J.T. and V.N. performed cytogenetic analysis; J.T. aided in manuscript preparation; R.K.R. provided NGS data; B.K.M. and M.L. performed molecular studies; J.M. was involved in design of the study and editing of the manuscript; and and H.K. and A.D. performed statistical analysis.
Conflict-of-interest disclosure: R.K.R. has received consulting fees from Incyte corporation, Celgene Corporation, Agios Pharmaceuticals, Apexx Oncology, and Jazz Pharmaceuticals; and has received research funding from Constellation Pharmaceuticals, Incyte Corporation, and Stemline Therapeutics. J.M. receives research funding from Incyte, Roche, Novartis, Celgene, Promedior, CTI Biopharma, Janssen, and Merck paid to the institution; and receives honorarium for participation in clinical trial steering committee and scientific advisory boards from Celgene, Roche, Inycte, and CTI Biopharma.The remaining authors declare no competing financial interests.
Correspondence: Vesna Najfeld, The Tisch Cancer Institute, Departments of Pathology and Medicine, 1425 Madison Ave, Box 1622, New York, NY 10029; e-mail: vesna.najfeld@mssm.edu.

