

Key Points
HDAC6 expression is upregulated in CLL patient samples, cell lines, and euTCL1 mouse models compared with normal controls.
Our results demonstrate therapeutic efficacy of selective HDAC6 inhibition in preclinical models, suggesting a rationale for treatment.

Abstract
Although the treatment paradigm for chronic lymphocytic leukemia (CLL) is rapidly changing, the disease remains incurable, except with allogeneic bone marrow transplantation, and resistance, relapsed disease, and partial responses persist as significant challenges. Recent studies have uncovered roles for epigenetic modification in the regulation of mechanisms contributing to malignant progression of CLL B cells. However, the extent to which epigenetic modifiers can be targeted for therapeutic benefit in CLL patients remains poorly explored. We report for the first time that expression of epigenetic modifier histone deacetylase 6 (HDAC6) is upregulated in CLL patient samples, cell lines, and euTCL1 transgenic mouse models compared with HDAC6 in normal controls. Genetic silencing of HDAC6 conferred survival benefit in euTCL1 mice. Administration of isoform-specific HDAC6 inhibitor ACY738 in the euTCL1 aging and adoptive transfer models deterred proliferation of CLL B cells, delayed disease onset via disruption of B-cell receptor signaling, and sensitized CLL B cells to apoptosis. Furthermore, coadministration of ACY738 and ibrutinib displayed synergistic cell kill against CLL cell lines and improved overall survival compared with either single agent in vivo. These results demonstrate for the first time the therapeutic efficacy of selective HDAC6 inhibition in preclinical CLL models and suggest a rationale for the clinical development of HDAC6 inhibitors for CLL treatment, either alone or in combination with Bruton tyrosine kinase inhibition.
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by the accumulation of mature CD5+ B cells in the peripheral blood, bone marrow, and secondary lymphoid tissues. The degree of somatic hypermutation of the immunoglobulin heavy chain variable (IGHV) region genes classify CLL into 2 main subtypes: mutated and unmutated.1 The unmutated subtype typically signifies a worse clinical prognosis than the mutated subtype, because it entails a more active proliferation of CLL B cells. Biological markers such as cytogenetic abnormalities, CD38, and ζ chain–associated protein kinase (ZAP) 70 expression can offer prognostic information for patients. At the molecular level, CLL B cells have been found to employ multiple strategies to support malignant progression, including aberrant signaling.2 Targeted therapies, such as ibrutinib, idelalisib, and venetoclax, have been developed to counter the effects of aberrant signaling in CLL B cells; however, patients may relapse, experience toxicities, develop resistant disease, or respond only partially.3 In these cases, rational combinations or novel therapeutic approaches are desirable.
Epigenetic reprogramming in CLL B cells has previously been described in several studies.4-6 These studies documenting DNA hypomethylation, dysregulated microRNAs, and histone modifications in CLL B cells have proven beneficial in identifying biomarkers and have offered a fresh perspective on therapeutic strategies. Our group has also reported findings establishing a role for epigenetic modifiers in altering CLL biology.7,8 Interestingly, various groups have reported elevated histone deacetylase (HDAC) expression in CLL B cells compared with HDAC expression in normal B cells and correlations of HDAC expression levels with prognostic factors.9-12 Several studies have also begun to explore the therapeutic potential of HDAC inhibitors for CLL treatment.13,14 However, the roles of individual HDACs in CLL biology have not been fully elucidated.
HDACs are divided into subfamilies according to their structural homologies, which are classes I, IIa, IIb, IV, and class III sirtuins. Although histones were originally classified as substrates of HDACs, these enzymes have also been found to directly regulate the activity of numerous nonhistone proteins.15 Class 2b HDAC6 can complex with nuclear proteins such as p300,16 HDAC11,17 Runx2,18 and LCoR19 to regulate transcriptional repression. Conversely, HDAC6 can deacetylate cytoplasmic proteins α-tubulin,20 cortactin,21,22 HSP90α,23 and GRP78,24 regulating cell motility, migration, division, and proteosomal degradation.25 Furthermore, HDAC6 has been explored in the context of various cancers. It was found to be upregulated in oral squamous cell carcinoma,26 ovarian carcinoma,27,28 and primary acute myeloid leukemia29 relative to nonmalignant controls. Preclinical studies have demonstrated that HDAC6 inhibitors display antitumor activity when used either alone or in combination with other agents to treat multiple myeloma,30,31 B-cell lymphoma,24,32 breast cancer,33,34 lung cancer,35,36 and melanoma.37,38 At present, HDAC6-selective inhibitors are in clinical trials to treat several solid tumor types, multiple myeloma, and lymphoma (registered at www.clinicaltrials.gov as #NCT03008018, #NCT02935790, #NCT20635061, #NCT01323751, and #NCT02091063).39 Finally, HDAC6 has been explored in the context of immune regulation.40,41 In the current study, we asked whether there is a role for HDAC6 in CLL B-cell pathobiology and explored the potential therapeutic value of targeting HDAC6 in preclinical CLL models.
Methods
CLL patient samples
Peripheral blood mononuclear cells were obtained from patients with CLL. All participants provided written institutional review board–approved informed consent for their blood to be used for research (#CR6_Pro00000316). Blood was collected at the H. Lee Moffitt Cancer Center and Research Institute (Tampa, FL).
In vivo studies
An HDAC6-deficient CLL murine model (referred to as euTCL1/HDAC6KO) was generated by crossing HDAC6KO and Eu-TCL1 (C57BL/6 background) mice.42,43 All euTCL1 and euTCL1/HDAC6KO mice were homozygous for T-cell leukemia 1 (TCL1) gene, and all mice harboring HDAC6KO were homozygous for the knockout as confirmed by genotyping. For the accelerated CLL model, several aged leukemic euTCL1 mice (aged leukemic was defined as >9 months of age and showing >70% CD5+ B cells of total viable lymphocytes by flow analysis) were euthanized, and their splenocytes were pooled. Freshly obtained splenocytes were then resuspended in phosphate-buffered saline and adoptively transferred via tail vein into 6- to 8-week-old C57BL/6 wild-type (WT) mice at 25 × 106 splenocytes per mouse. In severe combined immunodeficient (SCID) mice, leukemic splenocytes were injected at 5 × 106 splenocytes per mouse (WT or SCID recipient animals were purchased from Charles River). CLL induction was confirmed at 3 weeks after adoptive transfer by high complete blood count and a significantly greater CD19+ B220+ CD5+ B lymphocyte population in peripheral blood than in peripheral blood from a healthy age-matched WT cohort. Groups were randomized before treatment. For survival analyses, mice were monitored until death or euthanasia resulting from disease symptoms such as lethargy, difficulty moving, lack of grooming, and enlarged spleen and/or lymph nodes. Mice were kept in pathogen-free conditions and handled in accordance with Guidelines for Animal Experiments requirements.
Cell culture
Cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 1% nonessential amino acids, and MycoZap (Lonza). Cell lines were authenticated before use in these assays. Cells were incubated in 5% carbon dioxide at 37°C for the duration of assays.
Flow cytometry
For murine CLL immunophenotyping, 100 µL of peripheral blood was obtained from submandibular bleeds, and fresh spleens were homogenized and resuspended in an equal volume of flow cytometry (fluorescence-activated cell sorter) buffer before analysis. Cells were stained with indicated antibodies according to manufacturers’ dilutions for 1 hour at room temperature.
To complete phosphoflow, cells were stimulated and then fixed/permeabilized in 1% BD CytoFix for 15 minutes followed by 90% ice-cold methanol for 1 hour. Cells were stained with the phospho-specific antibody or isotype control for 1 hour before analysis in fluorescence-activated cell sorter buffer in the presence of Fc block.
For intracellular staining, cells were fixed/permeabilized using transcription factor fixation/permeabilization buffer set (BD Biosciences) in accordance with the manufacturer’s protocol. Cell cycling was achieved by fixing cells in ice-cold 80% ethanol dropwise and incubating them at −20°C overnight followed by Ki67 staining (BD Biosciences). Acquisition was performed on an LSRII Cytometer (BD Biosciences), and data were analyzed with FlowJo software v10.1 (Tree Star Inc.).
Statistical analyses
For normally distributed data, statistical significance of comparisons between data sets was determined by unpaired 2-tailed Student t test or 1-way analysis of variance followed by Tukey’s multiple comparisons test. For data that did not demonstrate normal distribution, Mann-Whitney U unpaired test or 1-way analysis of variance followed by Kruskal-Wallis test were used. Kaplan-Meier survival curves were compared using the log-rank test. Overall survival was defined as time from birth until death or euthanasia in the aged euTCL1 model or time from leukemic adoptive transfer until death or euthanasia in the accelerated model. For correlation studies, linear regression was performed on data sets. P < .05 was considered significant. All analyses were conducted using GraphPad Prism software v7.
Additional details can be found in supplemental Methods.
Results
Analysis of HDAC6 expression in CLL patient samples
We examined the expression of HDAC6 in peripheral blood samples from a cohort of 38 CLL donors and 13 healthy donors. Supplemental Table 1 displays clinical features of all CLL patients. PBMCs were gated on normal B cells (CD19+ CD5−) in healthy donors and CLL B cells (CD19+ CD5+) in CLL donors (supplemental Figure 1). HDAC6 protein expression was determined by flow cytometry and quantified by median fluorescence intensity. HDAC6 protein expression was found to be upregulated in CLL B cells compared with normal healthy donor B cells (Figure 1A). Correlation analyses demonstrated a weak positive association between HDAC6 protein expression and CD19+ CD5+ CLL B-cell percentages in CLL patient samples (Figure 1B). In addition, HDAC6 protein expression was higher among CD38+ patients than CD38− patients (Figure 1C). However, HDAC6 protein expression did not correlate with sex, age at diagnosis, ZAP70 status, IGHV region–mutated/unmutated status, or relapsed status.
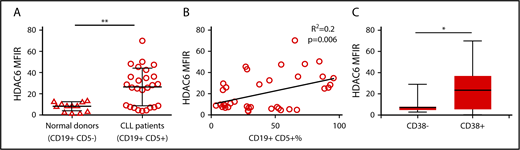
Analysis of HDAC6 expression in CLL patient samples. (A) Intracellular HDAC6 protein expression in CD19+ CD5− B cells from normal donors (n = 13) and CD19+ CD5+ B cells from CLL patient peripheral blood samples (n = 38), as determined by flow cytometry. (B) Correlation analyses showing relationship between HDAC6 protein expression and CLL B-cell percentages in CLL patient samples. (C) HDAC6 protein expression in CD38+ or CD38− CLL patient samples. Error bars correspond to standard deviations. *P < .05, **P < .005. MFIR, median fluorescence intensity ratio (value normalized to isotype control).
Analysis of HDAC6 expression in CLL patient samples. (A) Intracellular HDAC6 protein expression in CD19+ CD5− B cells from normal donors (n = 13) and CD19+ CD5+ B cells from CLL patient peripheral blood samples (n = 38), as determined by flow cytometry. (B) Correlation analyses showing relationship between HDAC6 protein expression and CLL B-cell percentages in CLL patient samples. (C) HDAC6 protein expression in CD38+ or CD38− CLL patient samples. Error bars correspond to standard deviations. *P < .05, **P < .005. MFIR, median fluorescence intensity ratio (value normalized to isotype control).
Genetic silencing of HDAC6 in the euTCL1 murine model
To study the role of HDAC6 in CLL B cells, we employed the euTCL1 transgenic murine model. This mouse model is genetically engineered to express the T-cell leukemia 1 (TCL1) gene in B cells, leading to development of a CLL-like phenotype at 6 to 9 months of age.43 At 6 months of age, euTCL1 mice demonstrated higher expression of HDAC6 protein in peripheral CD19+ B cells compared with age-matched WT mice (Figure 2A). Here, we generated an HDAC6-deficient euTCL1 mouse model by crossing euTCL1 mice with HDAC6KO mice. In euTCL1/HDAC6KO mice, HDAC6 protein levels were significantly decreased and TCL1 protein expression in B cells remained unchanged compared with euTCL1 mice (supplemental Figure 2). We also detected increased acetylation of α-tubulin in HDAC6-deficient mice,20 suggesting that HDAC6 was functionally impaired (supplemental Figure 2). Age-matched cohorts of euTCL1 and euTCL1/HDAC6KO mice were observed for survival alongside WT and HDAC6KO controls. Interestingly, the euTCL1/HDAC6KO cohort demonstrated increased overall survival (median survival, 375 days) compared with euTCL1 controls (median survival, 318.5 days; Figure 2B). However, euTCL1/HDAC6KO mice eventually succumbed to leukemia.
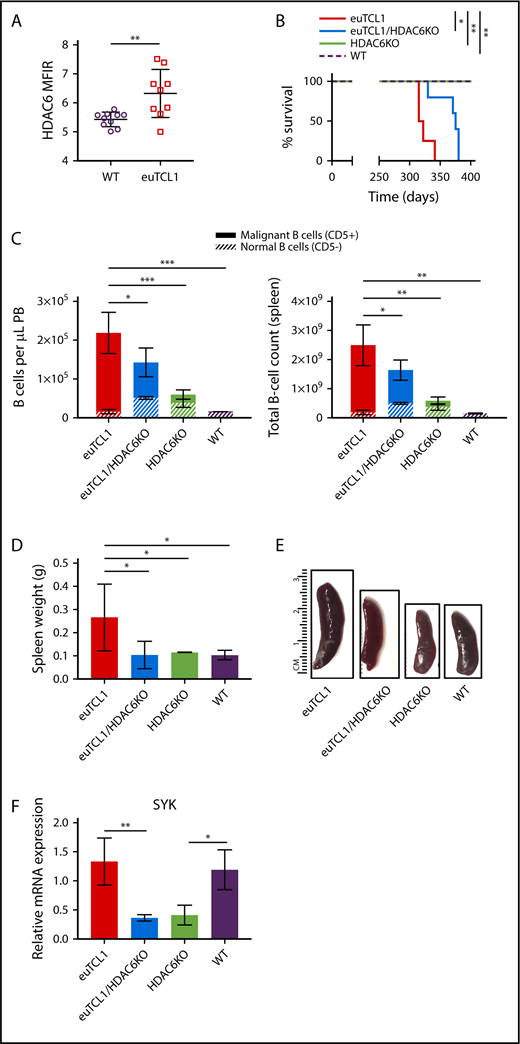
Genetic silencing of HDAC6 in euTCL1 mice. (A) Intracellular HDAC6 protein expression in CD19+ B220+ B cells from WT or euTCL1 peripheral blood (PB), as determined by flow cytometry (n = 9 per group). (B) Overall survival of indicated groups of mice (n = 5 per group), representing results of 3 independent experiments. (C) CLL burden quantified in PB or spleen by flow cytometry (n = 5 per group), representing results of at least 3 independent experiments. (D) Compilation of spleen weight data at the end of the study (n = 3 per group). (E) Representative spleens from each group indicated. (F) Spleen tyrosine kinase (SYK) messenger RNA (mRNA) expression in aged mice (n = 3 per group) normalized to 18s control gene. *P < .05, **P < .005, ***P < .0005.
Genetic silencing of HDAC6 in euTCL1 mice. (A) Intracellular HDAC6 protein expression in CD19+ B220+ B cells from WT or euTCL1 peripheral blood (PB), as determined by flow cytometry (n = 9 per group). (B) Overall survival of indicated groups of mice (n = 5 per group), representing results of 3 independent experiments. (C) CLL burden quantified in PB or spleen by flow cytometry (n = 5 per group), representing results of at least 3 independent experiments. (D) Compilation of spleen weight data at the end of the study (n = 3 per group). (E) Representative spleens from each group indicated. (F) Spleen tyrosine kinase (SYK) messenger RNA (mRNA) expression in aged mice (n = 3 per group) normalized to 18s control gene. *P < .05, **P < .005, ***P < .0005.
To determine the effect of HDAC6 silencing on CLL burden, 9-month-old mice were euthanized for the collection of spleens and peripheral blood. We quantified tumor burden by gating on CD3− CD19+ B220Lo-Hi CD5+ B cells (supplemental Figure 3). The total B-cell number (CD19+ B220Lo-Hi) was lower in the euTCL1/HDAC6KO group than the euTCL1 group, in both peripheral blood and spleen compartments. In addition, the ratio of CD5+ B cells (defined as malignant) to CD5− B cells (defined as normal) was reduced in the euTCL1/HDAC6KO group compared with the euTCL1 group. The reduction in malignant/normal B-cell ratios was evident in both peripheral blood and spleen (Figure 2C). Finally, splenomegaly was reduced in euTCL1/HDAC6KO mice compared with euTCL1 controls (Figure 2D-E). Genetic silencing of HDAC6 therefore slowed down the accumulation of malignant B cells in vivo, resulting in lower CLL tumor burden over time and improved overall survival rates.
RNA sequencing of HDAC6-deficient murine CLL B cells
Next, we investigated transcriptional changes related to disease progression in HDAC6-deficient CLL B cells. RNA sequencing was performed on B cells isolated from spleens of young (age 3 months) and aged (age 9 months) cohorts of euTCL1, euTCL1/HDAC6KO, HDAC6KO, and WT mice. Gene expression patterns in young mice differed from those in aged mice. Hierarchical clustering demonstrated that gene expression was most dissimilar between euTCL1 and euTCL1/HDAC6KO in both young and aged groups, suggesting that silencing of HDAC6 initiated widespread transcriptional changes in the CLL setting (supplemental Figure 4). In euTCL1 mice, CLL progresses as the mice age. Therefore, gene expression in aged euTCL1 may depend heavily on both the aging process and disease progression. Consequently, young and aged WT and HDAC6KO mice were compared to identify and eliminate genes that were differentially expressed because of the aging process.
Supplemental Figure 5 describes the analyses of the changed genes uniquely regulated in euTCL1/HDAC6KO mice compared with euTCL1 mice as a consequence of slower disease progression. The Venn diagram in supplemental Figure 5 depicts the group of 334 disease-related genes that were uniquely regulated in euTCL1/HDAC6KO mice compared with euTCL1 mice. Gene ontology analysis was performed on these 334 genes. A majority of the 50 pathways corresponded to immune-related signaling (supplemental Table 2). SYK was common among many of these pathways and was 1 of the most significantly changed genes within the focus group. It was confirmed by quantitative real-time polymerase chain reaction that SYK was transcriptionally downregulated in euTCL1/HDAC6KO and HDAC6KO mice compared with euTCL1 and WT mice, respectively (Figure 2F). Furthermore, it was confirmed that SYK protein was downregulated in euTCL1/HDAC6KO B cells compared with euTCL1 B cells (supplemental Figure 5). Interestingly, SYK is involved in transduction of BCR signaling, which is required for the survival of CLL B cells.2 Given the importance of SYK to BCR signaling, this result suggested to us that transcriptional regulation of SYK might potentially result in changes to downstream BCR signaling in CLL B cells lacking HDAC6.
Antitumor efficacy of ACY738 in murine CLL
Considering the results described, we asked whether selective HDAC6 inhibition could be harnessed for therapeutic benefit in CLL. First, orally bioavailable selective HDAC6 inhibitor ACY738 was incorporated into chow and fed to WT mice at levels equivalent to a final concentration of 25 mg/kg per day. At this concentration, ACY738 was detected in plasma (Figure 3A), and acetylation of α-tubulin was increased in the ACY738-treated group compared with a group fed chow (vehicle) alone (Figure 3B), confirming reduction of HDAC6 enzymatic activity with ACY738 oral treatment. ACY738-treated WT mice did not lose weight or show other signs of drug-related toxicities (data not shown). Percentage of normal CD19+ B cells, percentage of CD3+ T cells, CD4/CD8 ratio, and lymphocyte activation in spleen were not significantly affected after 1 month of continuous ACY738 treatment (Figure 3C).
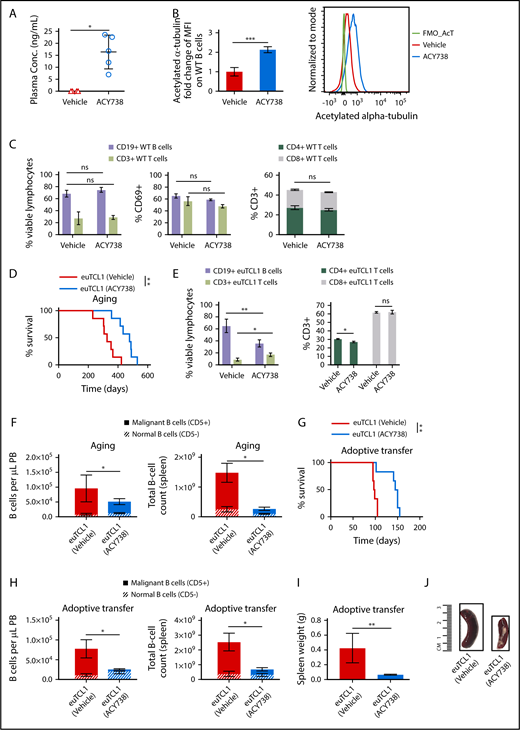
In vivo activity of selective HDAC6 inhibitor ACY738 in WT and CLL mice. Peripheral blood (PB) and spleens were collected from WT mice after 1 month of being fed vehicle (n = 4) or treated with ACY738 orally at 25 mg/kg (n = 5). (A) Plasma was separated by centrifugation from whole blood, and presence of ACY738 was detected by high-performance liquid chromatography. (B) Acetylation level of α-tubulin was quantified via flow cytometry in PB CD19+ B cells from indicated mice (n = 4 per group). (C) Characterization of immune subsets and activation status from splenocytes (n = 4 per group). (D) Overall survival for euTCL1 aging (n = 7 per group) mice fed vehicle only or treated with ACY738. Data are representative of 3 independent experiments. (E) Characterization of immune subsets from splenocytes derived from aging euTCL1 mice treated with vehicle or ACY738 orally (n = 7 per group). (F) CLL burden was quantified in aging euTCL1 mice fed vehicle only or treated with ACY738 for a duration of 3 months (n = 6 per group). Data were compiled from 2 independent experiments. (G) Overall survival for adoptive transfer euTCL1 mice (n = 6 per group) fed vehicle only or treated with ACY738. Data are representative of 3 independent experiments. (H) CLL burden was also quantified in adoptive transfer CLL mice (n = 8 per group) fed vehicle only or treated with ACY738 for a 12-week duration, and results are representative of 5 independent experiments. (I) Compilation of adoptive transfer CLL mice spleen weights after 12 weeks of indicated treatments (n = 6 per group). (J) Representative spleens from indicated groups. Error bars correspond to standard errors of the mean. *P < .05, **P < .005, ***P < .0005, compared with vehicle-only controls. conc, concentration; MFI, median fluorescence intensity; ns, not significant.
In vivo activity of selective HDAC6 inhibitor ACY738 in WT and CLL mice. Peripheral blood (PB) and spleens were collected from WT mice after 1 month of being fed vehicle (n = 4) or treated with ACY738 orally at 25 mg/kg (n = 5). (A) Plasma was separated by centrifugation from whole blood, and presence of ACY738 was detected by high-performance liquid chromatography. (B) Acetylation level of α-tubulin was quantified via flow cytometry in PB CD19+ B cells from indicated mice (n = 4 per group). (C) Characterization of immune subsets and activation status from splenocytes (n = 4 per group). (D) Overall survival for euTCL1 aging (n = 7 per group) mice fed vehicle only or treated with ACY738. Data are representative of 3 independent experiments. (E) Characterization of immune subsets from splenocytes derived from aging euTCL1 mice treated with vehicle or ACY738 orally (n = 7 per group). (F) CLL burden was quantified in aging euTCL1 mice fed vehicle only or treated with ACY738 for a duration of 3 months (n = 6 per group). Data were compiled from 2 independent experiments. (G) Overall survival for adoptive transfer euTCL1 mice (n = 6 per group) fed vehicle only or treated with ACY738. Data are representative of 3 independent experiments. (H) CLL burden was also quantified in adoptive transfer CLL mice (n = 8 per group) fed vehicle only or treated with ACY738 for a 12-week duration, and results are representative of 5 independent experiments. (I) Compilation of adoptive transfer CLL mice spleen weights after 12 weeks of indicated treatments (n = 6 per group). (J) Representative spleens from indicated groups. Error bars correspond to standard errors of the mean. *P < .05, **P < .005, ***P < .0005, compared with vehicle-only controls. conc, concentration; MFI, median fluorescence intensity; ns, not significant.
Next, groups of aging euTCL1 mice were treated with ACY738 or were fed vehicle alone, beginning at 3 months of age until either death or euthanasia because of disease symptoms. Similar to HDAC6-deficient euTCL1 mice, ACY738-treated euTCL1 mice displayed significantly longer overall survival compared with mice fed vehicle only (Figure 3D). Figure 3E displays the gross changes in percentage of B and T cells observed in the periphery of these mice. Specifically, tumor burden and malignant/normal B-cell ratios were decreased in peripheral blood and spleen samples after 3 months of treatment (Figure 3F). To extend these results, this experiment was replicated in the adoptive transfer euTCL1 model. In this model, leukemic euTCL1 splenocytes were injected into 6- to 8-week-old WT mice to establish CLL disease within 3 weeks of engraftment. Mice were then randomized to ACY738 or vehicle groups. ACY738 treatment significantly prolonged overall survival in this model as well (Figure 3G). Finally, in mice euthanized at the end of the experimental period (12 weeks after adoptive transfer), onset of CLL was delayed, and splenomegaly was reduced in the ACY738 group (Figure 3E-F) compared with the vehicle-only group (Figure 3H-J). Taken together, these data suggest that pharmacological inhibition of HDAC6 with ACY738 resulted in a therapeutic benefit in our CLL models.
ACY738 treatment alters proliferative capacity and sensitivity to apoptosis in euTCL1 B cells
To investigate the direct effects of pharmacological HDAC6 inhibition on CLL B cells, euTCL1 splenocytes were injected into SCID mice, which intrinsically lack B and T cells. Mice were randomized into 2 groups treated with ACY738 or fed vehicle only. Rapid expansion of adoptively transferred euTCL1 B cells was detected in peripheral blood 7 days after transfer. However, oral ACY738 treatment significantly attenuated the expansion of malignant B cells over time (Figure 4A). This led us to hypothesize that ACY738 may directly affect intrinsic survival signals of malignant B cells, creating a cytostatic effect and therefore a more indolent, less aggressive disease. To test this hypothesis, all animals were euthanized after 21 days of treatment, and B cells were isolated from splenocytes by negative selection. First, the proliferative status of B cells was measured by Ki67 staining, and as expected, it was significantly reduced in the ACY738 treatment group compared with the vehicle-only group (Figure 4B). In addition, isolated B cells from these mice were cultured ex vivo with lipopolysaccharide for 72 hours. B cells from ACY738-treated mice were found to be more sensitive to lipopolysaccharide-induced apoptosis than B cells in untreated mice, as measured by annexin V and viability staining (Figure 4C-D).
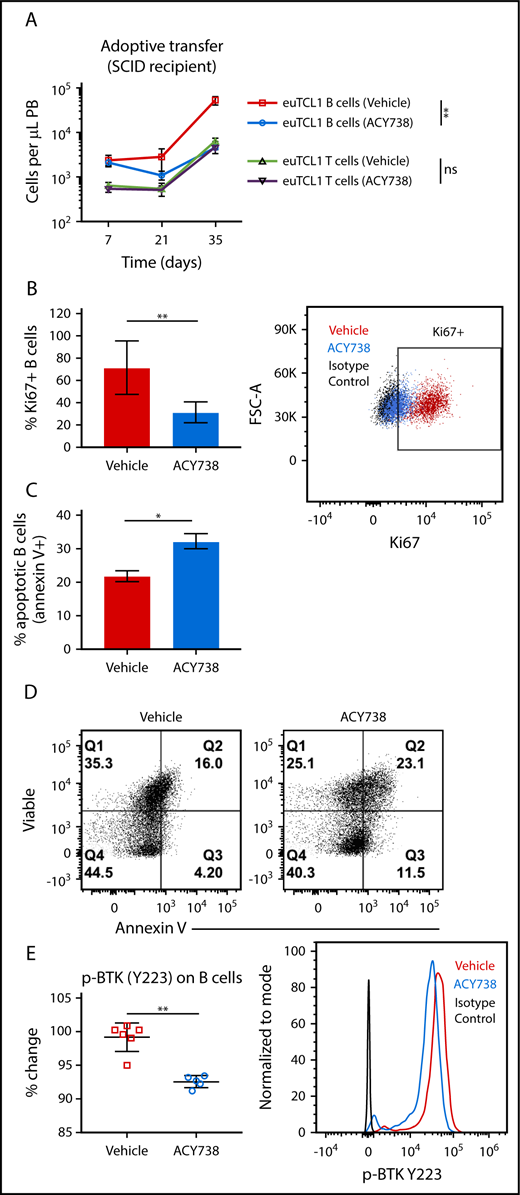
Effect of in vivo ACY738 treatment on euTCL1 B cells. (A) euTCL1 B cells were injected into SCID mice, which were then fed vehicle only or treated with oral ACY738 (n = 6 per group). Tumor burden was quantified in peripheral blood at the time points indicated. Data are representative of 2 independent experiments. (B) Total B cells were isolated from spleens of immunocompromised adoptive transfer CLL mice. Proliferative capacity was measured by Ki67 staining and flow cytometric analysis (n = 5 per group). (C) Isolated B cells were cultured with 1 μg/mL of lipopolysaccharide for 72 hours, and the percentage of apoptotic B cells was quantified by annexin V staining (n = 3 mice per group). (D) B cells were stimulated ex vivo with 10 μg/mL of anti-mouse immunoglobulin M, and phosphorylated Bruton tyrosine kinase (BTK; Y223) was analyzed by flow cytometry (vehicle-only group, n = 6 and ACY738-treated group, n = 5). Error bars correspond to standard errors of the mean. *P < .05, **P < .005.
Effect of in vivo ACY738 treatment on euTCL1 B cells. (A) euTCL1 B cells were injected into SCID mice, which were then fed vehicle only or treated with oral ACY738 (n = 6 per group). Tumor burden was quantified in peripheral blood at the time points indicated. Data are representative of 2 independent experiments. (B) Total B cells were isolated from spleens of immunocompromised adoptive transfer CLL mice. Proliferative capacity was measured by Ki67 staining and flow cytometric analysis (n = 5 per group). (C) Isolated B cells were cultured with 1 μg/mL of lipopolysaccharide for 72 hours, and the percentage of apoptotic B cells was quantified by annexin V staining (n = 3 mice per group). (D) B cells were stimulated ex vivo with 10 μg/mL of anti-mouse immunoglobulin M, and phosphorylated Bruton tyrosine kinase (BTK; Y223) was analyzed by flow cytometry (vehicle-only group, n = 6 and ACY738-treated group, n = 5). Error bars correspond to standard errors of the mean. *P < .05, **P < .005.
The malignant phenotype of CLL B cells depends on constitutive activation of molecular pathways that promote proliferation, resistance to apoptosis, and survival signals. BCR signaling is 1 of the most crucial pathways on which CLL B cells depend for intrinsic survival signals. Within this pathway, BTK is constitutively activated in CLL and is a critical kinase for CLL development and expansion.44 To examine whether activation status of this pathway was impaired, phosphorylation of BTK was measured after ex vivo stimulation with anti-mouse immunoglobulin M in the isolated B cells. Phosphorylation of BTK was found to be decreased in the ACY738 group compared with the vehicle-only group (Figure 4E). These results suggest that ACY738 treatment reduced BCR signaling in the malignant B cells, possibly disrupting downstream proliferation and antiapoptotic signals.
HDAC6 inhibition also alters proliferation and apoptosis in human CLL via downregulation of BCR signaling
To validate the relevance of these results in the human setting, 2 aggressive patient-derived CLL cell lines were treated with ACY738 in vitro to selectively inhibit HDAC6 (supplemental Figure 6A). Supplemental Figure 6B displays a heat map showing the selectivity of ACY738 for HDAC6 compared with other HDACs. The CLL cell lines OSU-CLL and Mec2 were found to express HDAC6 protein at higher levels than normal healthy human donor B-cell controls (Figure 5A). First, these cell lines were cultured with ACY738 or DMSO control. Growth kinetics were significantly delayed in ACY738-treated cells (Figure 5B; supplemental Figure 6C), with reduced cell-cycle progression and arrest in G1 phase (Figure 5C; supplemental Figure 6D). Additionally, CLL cells experienced increased, dose-dependent apoptosis after treatment with ACY738 relative to DMSO (Figure 5D-E; supplemental Figure 6E). To further examine the role of BCR signaling in altering CLL proliferation after HDAC6 inhibition, we measured the phosphorylation status of several BCR-signaling molecules after ACY738 treatment. Phosphorylation of several BCR-signaling molecules was dose dependently abrogated in CLL cell lines after ACY738 treatment compared with phosphorylation in DMSO-treated controls (Figure 5F; supplemental Figure 6F). Considering these results, we looked at MCL1 and BCL-2, transcription factors that mediate resistance to apoptosis in CLL cells.45 Both MCL1 and BCL-2 levels were reduced after 24 hours of culture with ACY738 in a dose-dependent manner (Figure 5G; supplemental Figure 6G).
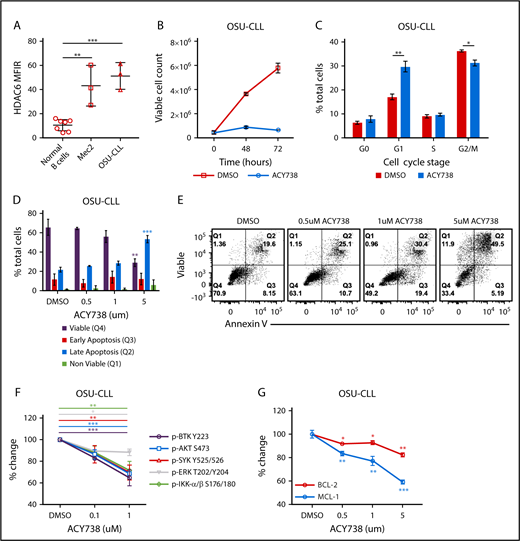
Effect of ACY738 treatment in human CLL cell lines. (A) HDAC6 protein expression quantified by flow cytometry in normal donor B cells (n = 5) and CLL cell lines; data were compiled from 3 independent experiments. (B) Growth kinetics of OSU-CLL in culture with ACY738 at 1 μM or vehicle for indicated period. (C) Cell-cycle analysis in OSU-CLL determined by Ki67/7AAD staining after 24 hours of indicated treatments. (D-E) Apoptosis was measured by annexin V/viable staining after treatment of OSU-CLL with ACY738 for 24 hours. (F) Phosphorylation of indicated molecules was measured after stimulation with 10 μg/mL of anti-human immunoglobulin M in CLL cell lines. (G) Expression of intracellular MCL1 and BCL-2 protein analyzed by flow cytometry, after incubating OSU-CLL with indicated concentrations of ACY738. All error bars correspond to standard deviations. *P < .05, **P < .005, ***P < .0005. DMSO, dimethyl sulfoxide.
Effect of ACY738 treatment in human CLL cell lines. (A) HDAC6 protein expression quantified by flow cytometry in normal donor B cells (n = 5) and CLL cell lines; data were compiled from 3 independent experiments. (B) Growth kinetics of OSU-CLL in culture with ACY738 at 1 μM or vehicle for indicated period. (C) Cell-cycle analysis in OSU-CLL determined by Ki67/7AAD staining after 24 hours of indicated treatments. (D-E) Apoptosis was measured by annexin V/viable staining after treatment of OSU-CLL with ACY738 for 24 hours. (F) Phosphorylation of indicated molecules was measured after stimulation with 10 μg/mL of anti-human immunoglobulin M in CLL cell lines. (G) Expression of intracellular MCL1 and BCL-2 protein analyzed by flow cytometry, after incubating OSU-CLL with indicated concentrations of ACY738. All error bars correspond to standard deviations. *P < .05, **P < .005, ***P < .0005. DMSO, dimethyl sulfoxide.
Synergistic activity of ACY738 and ibrutinib in vitro
On the basis of the antiproliferative and proapoptotic effects of HDAC6 inhibition, we hypothesized that combining HDAC6 and BTK (ibrutinib) inhibitors would be beneficial for CLL therapy. Indeed, combining ACY738 and ibrutinib demonstrated synergistic cell kill in both OSU-CLL and Mec2 cells in vitro (Figure 6A-B). To determine whether this was relevant in primary cells, we cultured CLL patient samples with ACY738, ibrutinib, or both. Single treatments decreased viability dose dependently; however, the combination further decreased viability compared with single treatments. Decreased viability was more noticeable in the unmutated patients, suggesting that patients with more proliferative disease may particularly benefit from this combination (Figure 6C).
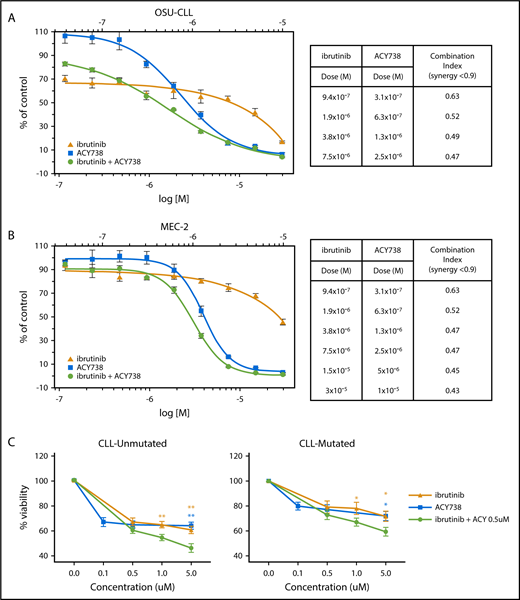
Combinatorial efficacy of ACY738 and ibrutinib in CLL cells in vitro. (A) CellTiter-Blue assay was performed to detect cell kill synergy in CLL cell lines treated with ACY738 and ibrutinib. (B) CellTiter-Blue assay was performed to detect cell kill in CLL patient peripheral B cells with unmutated or mutated IGVH status after 48 hours of indicated treatments (n = 8 patients per group). All error bars correspond to standard deviations. *P < .05, **P < .005.
Combinatorial efficacy of ACY738 and ibrutinib in CLL cells in vitro. (A) CellTiter-Blue assay was performed to detect cell kill synergy in CLL cell lines treated with ACY738 and ibrutinib. (B) CellTiter-Blue assay was performed to detect cell kill in CLL patient peripheral B cells with unmutated or mutated IGVH status after 48 hours of indicated treatments (n = 8 patients per group). All error bars correspond to standard deviations. *P < .05, **P < .005.
Combinatorial efficacy of ACY738 and ibrutinib in murine CLL in vivo
To assess the in vivo relevance of this drug combination, groups of adoptively transferred euTCL1 mice were treated with ACY738 alone, ibrutinib alone, or ACY738 in combination with ibrutinib or fed vehicle only from disease engraftment until death or euthanasia. Before these experiments, we confirmed that WT mice treated with ibrutinib via drinking water had exhibited reduced phosphorylation of BTK in isolated splenic B cells after only 3 days of ibrutinib treatment (supplemental Figure 7). First, longer overall survival was confirmed in both the ACY738-only and ibrutinib-only groups as compared with the vehicle group. However, the combined treatment further increased overall survival compared with either single-agent treatment group (Figure 7A). Median survival of the dual-treatment group was 194 days compared with 112.5 days for the ACY738-only group and 138.5 days for the ibrutinib-only group. When euthanized after 12 weeks, tumor burden and splenomegaly were observed to be lower in the dual-treatment group (Figure 7B-C) than in either single-agent group (Figure 7D-E). Mice receiving dual treatment did not overtly exhibit any symptoms associated with drug-related toxicities, such as weight loss or diarrhea (data not shown). These results suggest that dual inhibition of HDAC6 and BTK showed combinatorial antitumor efficacy against CLL in this murine model.
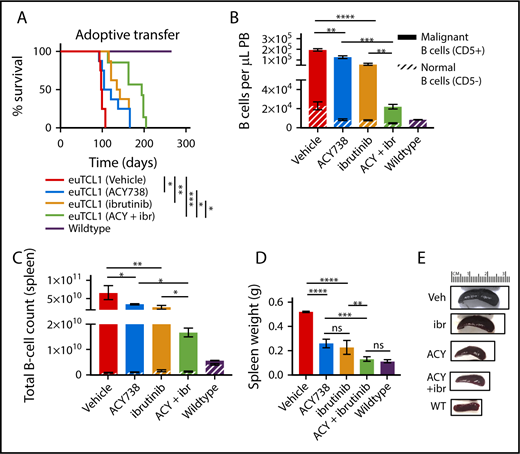
Combinatorial antitumor efficacy of ACY738 and ibrutinib treatment in CLL mice. (A) Overall survival data for adoptive transfer CLL mice treated as indicated until death or euthanasia (n = 8 per group), representative of 2 independent experiments. (B-C) CLL burden was quantified in peripheral blood or spleen after 12 weeks of indicated treatments; data are representative of 3 independent experiments. (D) Spleen weight at conclusion of experiment. (E) Representative spleens from indicated treatment groups are shown. Error bars correspond to standard errors of the mean. *P < .05, **P < .005, ***P < .0005, ****P < .00005.
Combinatorial antitumor efficacy of ACY738 and ibrutinib treatment in CLL mice. (A) Overall survival data for adoptive transfer CLL mice treated as indicated until death or euthanasia (n = 8 per group), representative of 2 independent experiments. (B-C) CLL burden was quantified in peripheral blood or spleen after 12 weeks of indicated treatments; data are representative of 3 independent experiments. (D) Spleen weight at conclusion of experiment. (E) Representative spleens from indicated treatment groups are shown. Error bars correspond to standard errors of the mean. *P < .05, **P < .005, ***P < .0005, ****P < .00005.
Discussion
In this study, we found that protein expression of HDAC6 was elevated in B cells of CLL patients compared with normal B-cell controls (Figure 1). Previously, Van Damme et al11 demonstrated that HDAC6 mRNA was found to be higher in CLL patient B cells compared with normal B-cell controls. In addition, HDAC6 mRNA expression was found to correlate with treatment-free survival but not with CD38, ZAP70, or IGVH status. In our study, HDAC6 protein expression was higher in CD38+ patients compared with CD38− patients. CD38+ CLL patients typically represent a population of intermediate- to high-risk patients. It is interesting to note that, in separate studies, both HDAC6 mRNA and protein were found to be elevated in CLL B cells. Our data also showed elevation of HDAC6 protein in CLL cell lines (Figure 5) and euTCL1 B cells (Figure 2) compared with normal controls. The discrepancies between these studies may be due to differences in methods. Protein level was measured via flow cytometry in the current study, whereas mRNA level was measured via quantitative real-time polymerase chain reaction in the study by Van Damme et al.11 In addition, cohort size and average CLL B-cell percentage present in the patient samples were different between the 2 studies. Altogether, our current observations and the prior literature suggest that HDAC6 expression may be a clinically relevant factor for CLL patients and warrant further investigation to determine whether HDAC6 may be able to regulate processes supporting the malignant progression of CLL B cells.
Our experiments showed that HDAC6 inhibition downregulated BCR signaling in murine and human CLL B cells, leading to defects in proliferation and sensitivity to apoptosis. This translated to delayed disease progression, lower tumor burden, and increased overall survival when HDAC6 was genetically silenced or pharmacologically inhibited in the euTCL1 model. Molecules such as BTK and AKT that are constitutively activated downstream of the BCR are particularly crucial to transduction of oncogenic signaling in CLL B cells.46-50 HDAC6 has been shown to regulate oncogenic mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K) signaling pathways in multiple cell types via deacetylation of cytoplasmic targets.51 In our experiments, we found no evidence of hyperacetylation of SYK, BTK, or AKT after HDAC6 inhibition in CLL cell lines (data not shown). However, RNA sequencing analyses demonstrated transcriptional regulation of several genes that may be involved in CLL progression. Of note, SYK was included in this list. Given the importance of SYK to BCR signaling transduction, this result indicated to us that HDAC6 might be acting in the nucleus to regulate the expression of this kinase, thereby affecting downstream BCR signaling. Although the exact mechanism is not yet clear, we have additional studies that are fully exploring this possibility.
Single-agent therapy with BCR inhibitors has met with clinical success; however, relapsed patients are emerging as a population with unmet needs. Characterization of mutations in BCR-signaling components, such as BTK (C481S) and/or PLC-2γ, has shown a role for these components in resistance to ibrutinib. Further characterization also found genetic alterations of TRAIL-R, EP300, MLL2, and EIF2A in ibrutinib-resistant patients. Mutations could be detected for up to 15 months before manifestation of clinical progression, and these patients were transitioned to alternative targeted agents with moderate success.52 A study of clonal evolution in 8 patients who were resistant to venetoclax (BCL-2 inhibitor) reported mutations in BTG1 and BRAF deletions affecting CDKN2A and amplification of CD274 (PD-L1).53 Other researchers have predicted that resistance to venetoclax may occur via upregulation of alternative antiapoptotic BCL family proteins. Resistance to idelalisib mechanisms (PI3Kδ inhibitors) have not yet been described in CLL patients but have been identified in other cancers and are predicted to occur, for example, via upregulation of class 1A PI3K.54 Combination approaches using ibrutinib are being tested in previously treated high-risk patients and in treatment-naïve patients (#NCT02301156, #NCT02420912, and #NCT02756897). Results reported after 6 months of a clinical trial testing ibrutinib plus venetoclax in relapsed/refractory (R/R) CLL showed high overall response rates, eradication of minimal residual disease, and manageable adverse events.55 Although results are promising, long-term follow-up has not been reported, and it is not known whether some of these patients may ultimately progress.
In conclusion, this study establishes for the first time the potential therapeutic value of pharmacological HDAC6 inhibition for CLL treatment. ACY738 and ibrutinib demonstrated greater cell kill than either single treatment in CLL B cells, particularly in patients with unmutated IGVH, and significant combinatorial antitumor efficacy in our preclinical CLL models. Despite improvements in care and the option of allogeneic bone marrow transplantation, CLL currently remains incurable and demonstrates a variable course with many R/R patients. In anticipation of the drawbacks of the currently available therapies, it is imperative to develop alternative inhibitors and rational combination approaches to cater to those patients who do not respond to or experience progression with available regimens. One strategy being explored to combat ibrutinib resistance is combination with immunotherapy or inhibitors whose antitumor efficacy results from a differentiated mechanism of action. Given our preclinical data, we believe selective HDAC6 inhibitors are promising candidates for this niche and are worthy of further testing either alone, in combination with BTK inhibitors, or as an alternative therapeutic agent for R/R patients.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors acknowledge Carlo Croce, who kindly provided the euTCL1 model, and the comparative medicine (vivarium) staff at the H. Lee Moffitt Cancer Center and Research Institute for their extended technical support of our project. The authors thank Sonya J. Smyk of Moffitt Cancer Center for editorial support. She was not compensated beyond her regular salary.
This work was supported by by a Leukemia and Lymphoma Society Translational Research Program grant and Acetylon Pharmaceuticals and in part by the Flow Cytometry and Molecular Genomics Core Facilities at the Moffitt Cancer Center and Research Institute, a National Institutes of Health, National Cancer Institute–designated comprehensive cancer center (P30-CA076292).
Authorship
Contribution: K.M. designed and conducted research, analyzed and interpreted data, and wrote the manuscript; J.J.P. designed and conducted research and analyzed data; A.A., S.D., R.F., and M.P.-S. conducted research; S.N.Q. and S.S.J. served as project advisers; A.V. and E.M.S., as experts in the field, advised on the data generated; and E.S. and J.P.-I. conceived and designed the research and revised the manuscript.
Conflict-of-interest disclosure: J.P.-I. and E.S. received research funding from Acetylon Pharmaceuticals, Inc., and subsequently from Celgene. The remaining authors declare no competing financial interests.
Correspondence: Javier Pinilla-Ibarz, Moffitt Cancer Center & Research, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: javier.pinilla@moffitt.org; and Eva Sahakian, Moffitt Cancer Center & Research Institute, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: eva.sahakian@moffitt.org.

