

Key Points
Passive immunization results in antigen-specific antigen modulation and AMIS.
AMIS related to antigen modulation does not alter the immune response to other foreign antigens present on transfused RBC.

Abstract
Alloantibodies developing after exposure to red blood cell (RBC) alloantigens can complicate pregnancy and transfusion therapy. The only method currently available to actively inhibit RBC alloantibody formation is administration of antigen-specific antibodies, a phenomenon termed antibody-mediated immune suppression (AMIS). A well-known example of AMIS is RhD immune globulin prophylaxis to prevent anti-D formation in RhD− individuals. However, whether AMIS is specific or impacts alloimmunization to other antigens on the same RBC remains unclear. To evaluate the specificity of AMIS, we passively immunized antigen-negative recipients with anti-KEL or anti-hen egg lysozyme (HEL) antibodies, followed by transfusion of murine RBC expressing both the HEL-ovalbumin-Duffy (HOD) and human KEL antigens (HOD × KEL RBC). Significant immunoglobulin G deposition on transfused HOD × KEL RBC occurred in all passively immunized recipients. Complement deposition and antigen modulation of the KEL antigen occurred on transfused RBC only in anti-KEL–treated recipients, whereas HEL antigen levels decreased only in the presence of anti-HEL antibodies. Western blot analysis confirmed the specificity of antigen loss, which was not attributable to RBC endocytosis and appears distinct for the 2 antigens. Specifically, removal of KEL was attenuated by clodronate treatment, whereas loss of HEL was unaffected by clodronate in vivo but sensitive to protease treatment in vitro. Antigen-specific modulation correlated with antigen-specific AMIS, with anti-KEL treated recipients forming antibodies to the HOD antigen and anti-HEL–treated recipients developing antibodies to the KEL antigen. Together, these results demonstrate that passively administered antibodies can selectively inhibit the immune response to a specific antigen.
Introduction
Antibodies to red blood cell (RBC) antigens can develop after exposure to RBC alloantigens during pregnancy or transfusion therapy. Such antibodies complicate transfusion therapy by causing hemolytic transfusion reactions, or pregnancies by causing hemolytic disease of the fetus and newborn.1-3 Additionally, alloantibodies to RBC antigens can decrease the therapeutic efficacy of transfused RBC and hinder finding compatible RBC for future transfusions.4,5 Excluding antigen-matching protocols to reduce alloimmunization risk,6,7 the sole therapeutic intervention currently available to prevent RBC alloantibody development is polyclonal RhD–immune globulin (RhIg).8 Prophylactic administration of RhIg to RhD− women successfully prevents the generation of de novo anti-RhD antibodies associated with pregnancy through a process termed antibody-mediated immune suppression (AMIS),9-13 whereby passively acquired antibody inhibits sensitization to a given antigen. Although AMIS is a successful clinical intervention, no such therapeutic options exist to prevent formation of other clinically significant non-RhD alloantibodies. This likely reflects both our inability to fully understand RhIg-related AMIS and variability in the mechanism and outcome of AMIS depending on the specific antigens or antibodies involved.14-20
Several mechanisms by which AMIS exerts its inhibitory effect have been postulated and tested, aided recently by the development of improved animal models. These include rapid induction of RBC clearance, antigen masking with steric hindrance of B-cell receptors, inhibition of B-cell responses through inhibitory Fcγ receptor engagement, and direct antigen modulation, which refers to removal of antigen from the cell surface and renders a cell antigen-negative as it persists in circulation.16,17,21,22 RBC clearance has been regarded as the primary mechanism of AMIS and occurs following passive immunization to some antigens, including RhD.23 However, recent evidence implicates antigen modulation as another mechanism by which AMIS can occur independently of RBC clearance.14,15 Previous reports supporting antigen modulation as a mechanism of AMIS were performed using murine models in which RBC express only 1 foreign antigen, so it remains unclear whether antigen modulation is a potential mechanism of AMIS in the context of multiple foreign RBC antigens expressed simultaneously, which is most often the case clinically.14,15,20,24,25
Previous observations suggest AMIS resulting from RBC clearance is nonspecific and inhibits sensitization to additional RBC alloantigens. For example, before RhIg availability, decreased rates of anti-RhD alloimmunization were recognized in RhD− mothers who were also ABO-incompatible with their babies.26-28 Another study in human volunteers found suppression of RhD sensitization in RhD− KEL− men exposed to RhD+ KEL+ RBC and passively immunized with anti-KEL immunoglobulin G (IgG).26 These observations contribute to the notion that AMIS is not antigen-specific and instead reflects rapid antibody-induced clearance of transfused cells before the adaptive immune response can detect foreign RBC antigens. More recent findings reported on the lower rates of human leukocyte antigen sensitization in previously pregnant RhD− women, who presumably received RhIg, compared with previously pregnant RhD+ women, again suggesting that AMIS may not be antigen-specific.29 Nevertheless, some women treated with RhIg do become alloimmunized to non-RhD fetal RBC antigens, highlighting conflicting evidence regarding whether AMIS can be antigen-specific and whether specificity depends on the antigens involved.30,31
Because the antigen-specific nature of AMIS has not been evaluated in the context of antigen modulation, we sought to clarify whether antigen modulation plays a role in AMIS when multiple foreign antigens are present on the RBC surface and whether this mechanism of AMIS is antigen-specific. In particular, we investigated whether antibody-specificity to 1 RBC antigen induces antigen-specific antigen modulation that results in antigen-specific AMIS. To address this, we generated transgenic mice expressing both the model hen egg lysozyme (HEL)–ovalbumin-human Duffy (HOD) antigen and the human KEL antigen on RBC (HOD × KEL RBC). We then transfused wild-type recipients, passively immunized with anti-KEL or anti-HEL antibodies, or phosphate-buffered saline (PBS) control, with HOD × KEL RBC. We assessed changes in the level of detectable antigen, as well as any impact of antigen-specific antibodies on the immune response to another antigen present on transfused RBC. Here we report that antigen-specific antibodies cause antigen-specific antigen modulation and result in antigen-specific AMIS on RBC bearing multiple foreign antigens.
Materials and Methods
Mice
All mice were housed in the Emory Division of Animal Resources and Husbandry facilities and treated in accordance with protocols approved by the Emory University Institutional Animal Care and Use Committee. HOD × KEL transgenic mice were generated as described previously.32,33 Wild-type C57BL/6 (WT) recipient mice were purchased from Charles River (Montreal, QC, Canada). Male and female mice were used between 8 and 12 weeks of age.
Passive immunization and transfusion
WT recipients were passively immunized with either anti-KEL polyclonal antibody (50 μL of sera from KEL-immunized mice generated as previously described)15 or a combination of 2 anti-HEL monoclonal antibodies (1.25 μg/mL each of clones 2F4 and 4B7, both from Bio X Cell, West Lebanon, NH) previously shown to induce AMIS to the HEL antigen34 or received PBS control by tail vein injection before transfusion. In some experiments, mice were depleted of macrophages by pretreatment with 500 μL clodronate or empty control liposomes (Liposoma, Amsterdam, The Netherlands) via tail vein injection 48 and 24 hours before transfusion, as done previously.35,36 For transfusions, HOD × KEL or WT RBC were collected into acid citrate dextrose (ACD, BD, Franklin Lakes, NJ) and washed 3 times in PBS. HOD × KEL+ and HOD × KEL− RBC were labeled with 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate and 3,3′-dioctadecyloxacarbocyanine perchlorate, respectively, as previously described.20,35 Each recipient was transfused a total volume of 300 μL, including 50 μL packed HOD × KEL RBC and 50 μL packed WT RBC in PBS, the adjusted equivalent of 1 human RBC unit.
Staining and antibody identification by flow cytometry
Platelets and splenocytes were collected from WT and HOD × KEL mice as previously described.37 Following isolation, platelets and splenocytes were stained with lineage-specific markers, CD41 and CD45, respectively (BD) as previously described.37 RBC were collected from recipients either before transfusion for characterization of HOD and KEL expression or following transfusion, at 1 hour, 2 hours, and 24 hours posttransfusion, and stained for flow cytometry as previously described.24,38-40 Briefly, RBC were stained with APC anti-mouse IgG (Jackson Immunoresearch, West Grove, PA) for assessment of bound antibody, or with biotin anti-mouse complement component 3 (C3; Cedarlane, Burlington, NC) for assessment of total complement or biotin anti-mouse C3b (Cedarlane) for detection of active complement, followed by streptavidin APC (BD, Franklin Lakes, NJ). KEL antigen was measured using anti-KEL polyclonal antibody followed by APC anti-mouse IgG secondary (Jackson Immunoresearch). HEL antigen was measured using anti-HEL monoclonal antibodies followed by secondary anti-mouse IgG APC (Jackson Immunoresearch). Both KEL and HEL antigens were measured as the percentage of KEL or HEL antigen observed in antibody-treated groups compared with control PBS-treated mice. Ter119 was measured using APC anti-mouse Ter119 (BD). To assess for the development of de novo anti-KEL and anti-HOD IgM and IgG antibodies, serum was flow cross-matched as previously described.15,25,37,41,42 All samples were run on a BD FACSCalibur flow cytometer and analyzed using FlowJo software (Tree Star Inc. Ashland, OR).
Immunofluorescent super-resolution microscopy
One milliliter of a 1:10 dilution of poly-l-lysine in PBS (Sigma 0.01% stock solution) was added to each 15-mm diameter glass coverslip. Following 1 hour incubation, coverslips were washed 3 times with PBS and then air-dried. Anti-HEL antibody was directly labeled using Alexa Fluor 488 NHS Ester (succinimidyl ester, ThermoFisher Scientific, Waltham, MA). HOD × KEL RBC were stained with polyclonal anti-KEL antibody followed by anti-mouse IgG-Alexa 555 (Invitrogen, Carlsbad, CA). This was followed by staining with directly labeled anti-HEL antibody. Stained HOD × KEL RBC were added to coverslips and incubated for 30 minutes on ice, followed by fixation with 1 mL of 1% paraformaldehyde in PBS overnight. Imaging was performed with a Nikon N-SIM super-resolution microscope.
Western blot analysis
Isolated RBC were lysed in 5 mmol/L sodium phosphate (pH 7.5) with protease inhibitor cocktail (Sigma Roche, St. Louis, MO). RBC ghosts were collected by centrifugation and washed with lysis buffer until translucent, as previously described.15,42 Western blot antibodies included anti-KEL antibody (Abcam, clone MM0435-12X3; diluted 1:250 in blocking buffer), rabbit polyclonal anti-HEL antibody (Abcam, ab391; 1:5,000), and goat anti-rabbit horseradish peroxidase (Abcam, ab205718; 1:10 000) for detection. Mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (clone: GA1R, ThermoFisher Scientific; 1:1,000), followed by goat anti-mouse IgG1 horseradish peroxidase (Bethyl Laboratories, Montgomery, TX; 1:10 000) was applied after stripping membranes with 25 mM glycine-HCl with 1% sodium dodecyl sulfate, pH 2. Membranes were developed using HyGlo enhanced chemiluminescence (Denville Scientific Inc., Holliston, MA).
Antigen assessment in vitro
HOD × KEL RBC were incubated with various dilutions of either polyclonal anti-KEL sera or combination anti-HEL antibodies, or PBS control, for 60 minutes at 37°C as indicated. Saturating concentrations of polyclonal anti-KEL sera (1:16 dilution) and anti-HEL monoclonal antibodies (1.25 μg/mL of each clone, 2F4 and 4B7) were used in RBC endocytosis assays and protease-treatment assays. For RBC endocytosis assays, antibody-sensitized cells were washed twice and then incubated at 4°C or 37°C in complete media (RPMI 1640 Medium, Sigma; containing 10% fetal calf serum and penicillin-streptomycin, Thermo Fisher Scientific) for 2 or 24 hours. For protease treatment, sensitized HOD × KEL RBC were washed and incubated for 30 minutes at 37°C with decreasing concentrations of Pronase (Sigma) starting from 5 μg/mL. Following treatment, RBC were washed and stained for KEL, HEL and Ter119 antigens as described previously.
Statistics
Statistical analysis was performed on in vitro experiments conducted in triplicate and in vivo experiments using 5 to 8 mice per group, as stated in the figure legends. Significance was determined using GraphPad Prism (GraphPad Software, San Diego, CA) and defined as P < .05. Analysis of RBC survival was performed using 2-way analysis of variance (ANOVA) with the Dunnett multiple comparisons test. Unpaired Student t test was used to compare results between 2 groups at a single time point. One-way ANOVA with Tukey multiple comparisons test was used to compare all other results with 3 or more groups, as noted in the figure legends.
Results
To determine whether AMIS demonstrates antigen-specific restriction, we generated mice whose RBC dually express 2 foreign antigens, HOD and KEL, as previously reported (Figure 1A).43 RBC ghosts generated from WT or HOD × KEL RBC and subjected to western blot analysis–confirmed expression of both HOD and KEL antigens only on HOD × KEL RBC (Figure 1B). Expression of HOD and KEL individually, as well as dual staining of HOD and KEL together, was restricted to RBC (Figure 1C), with no expression on other cell populations, such as platelets (Figure 1D) or white blood cells (Figure 1E). The presence of HOD and KEL antigens was further confirmed by super-resolution microscopy, which demonstrated dual expression of HOD and KEL on HOD × KEL RBCs with distinct localization (Figure 1F).
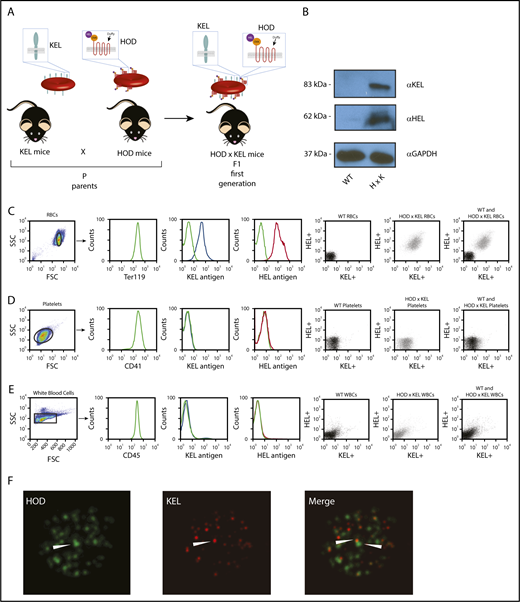
Characterization of HOD × KEL mouse model. (A) Transgenic mice expressing HOD or KEL on their RBC were bred to generate HOD × KEL mice. (B) KEL and HEL expression on WT and HOD × KEL RBC ghosts were determined by western blot analysis. Expression of KEL and HEL was determined by flow cytometry on WT and HOD × KEL RBCs (C), platelets (D), or white blood cells (WBCs) (E), individually and by dual staining of KEL and HEL together. (F) Anti-KEL and anti-HEL binding to HOD × KEL RBC was assessed by super-resolution microscopy (magnification ×100).
Characterization of HOD × KEL mouse model. (A) Transgenic mice expressing HOD or KEL on their RBC were bred to generate HOD × KEL mice. (B) KEL and HEL expression on WT and HOD × KEL RBC ghosts were determined by western blot analysis. Expression of KEL and HEL was determined by flow cytometry on WT and HOD × KEL RBCs (C), platelets (D), or white blood cells (WBCs) (E), individually and by dual staining of KEL and HEL together. (F) Anti-KEL and anti-HEL binding to HOD × KEL RBC was assessed by super-resolution microscopy (magnification ×100).
Because antibody binding and saturation levels may differ in vitro vs in vivo, we next determined whether saturation of HOD × KEL RBC occurred in vitro and whether this saturation reflected antibody binding in vivo. To accomplish this, we incubated HOD × KEL RBC with anti-KEL or anti-HEL antibody in vitro. We also exposed WT mice to HOD × KEL RBC following passive immunization with anti-KEL or anti-HEL antibodies and assessed antibody binding to transfused RBC by flow crossmatch (supplemental Figure 1A). Saturation of HOD × KEL RBC by anti-KEL antibodies occurred over a wide range of anti-KEL dilutions (supplemental Figure 1B). Importantly, similar saturation of HOD × KEL RBC was observed in vivo compared with in vitro binding of antibodies to HOD × KEL RBC from the sera of anti-KEL immunized mice to a saturating concentration of anti-KEL antibody (supplemental Figure 1C). We also observed saturation of HOD × KEL RBC by anti-HEL antibodies in vitro over a wide range of concentrations (supplemental Figure 1D), with saturation of HOD × KEL RBC likewise occurring in vivo, when we compared the binding of antibodies from the sera of anti-HEL passively immunized mice to a saturating concentration of anti-HEL antibody in vitro (supplemental Figure 1E). These results suggest that saturation of HOD × KEL RBC can occur both in vitro and in vivo using either anti-KEL or anti-HEL antibodies.
Because steric hindrance may affect antibody binding in a dual antigen model, we also tested whether antibody binding to 1 antigen interfered with antibody binding to the other antigen (supplemental Figure 1F). HOD × KEL RBC were incubated with various dilutions of anti-KEL sera before anti-HEL antibody incubation to see whether anti-KEL antibody binding affects the ability of anti-HEL antibodies to detect the HEL antigen (supplemental Figure 1F). HEL antigen was detected by anti-HEL antibodies on HOD × KEL RBC over a wide range of anti-KEL dilutions, suggesting that anti-KEL binding does not sterically hinder the ability of anti-HEL antibody to bind HOD × KEL RBC (supplemental Figure 1G). These results suggest that although HOD and KEL are expressed on the same RBC, antibody binding to 1 RBC antigen does not interfere with antibody binding to the other RBC antigen.
Antibody and complement deposition on transfused HOD × KEL RBC
To examine the specificity in the immune response to either RBC antigen when they are both expressed on the same RBC, we tracked HOD × KEL RBC posttransfusion.20,35 WT mice, passively immunized with anti-KEL or anti-HEL antibodies or injected with PBS control, were transfused with a 1:1 mixture of HOD × KEL and WT RBC (Figure 2A). Antibody deposition on transfused HOD × KEL cells was determined using a direct antiglobulin test by flow cytometry at 1, 2, and 24 hours posttransfusion. Recipients that received anti-KEL or anti-HEL antibodies showed bound antibody on the surface of transfused HOD × KEL RBC, unlike the PBS control group, which demonstrated no antibody binding (Figure 2B). The degree of antibody deposition appeared to undergo a significant initial decrease by 1 hour posttransfusion in groups receiving either anti-KEL or anti-HEL antibodies when compared with the maximum antibody binding detected before transfusion (Figure 2B; supplemental Figure 1), and displayed a gradual reduction over time thereafter (Figure 2B). Together, these data reflect the ability of these antibodies to selectively bind to their target antigen on transfused HOD × KEL RBC in vivo.
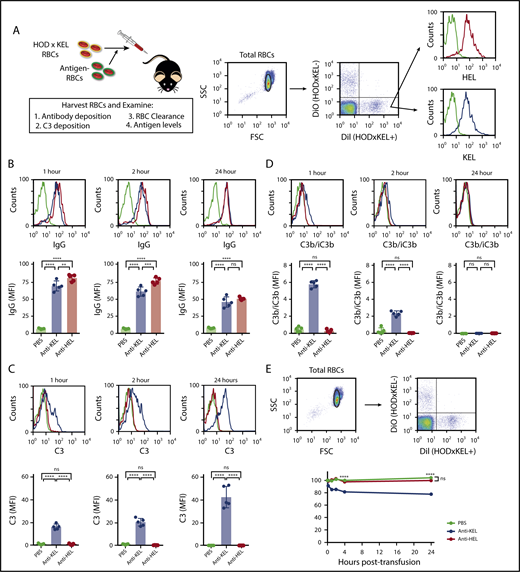
Antibody and complement deposition is detectable on HOD × KEL RBC posttransfusion, with clearance of HOD × KEL RBC occurring in anti-KEL immunized recipients. (A) HOD × KEL RBC were labeled before transfusion into PBS (no antibody), anti-KEL–, or anti-HEL–treated WT mice. (B) IgG deposition was measured by direct antiglobulin test at 1, 2, and 24 hours posttransfusion. Total C3 (C) and active complement (C3b/iC3b) (D) was measured at 1, 2, and 24 hours posttransfusion. (E) RBC survival of HOD × KEL+ compared with HOD × KEL− RBC at various times posttransfusion. Means ± standard deviation (SD) shown. (A-D) ****P < .0001, ***P < .001, **P < .01, and not significant (ns) by 1-way ANOVA with Tukey multiple comparison test. (E) ****P < .0001 and ns by 2-way ANOVA with Dunnett multiple comparisons test. Data shown include 5 mice per group and are representative of 3 independent experiments. DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; DiO, 3,3′-dioctadecyloxacarbocyanine perchlorate; FSC, forward scatter; MFI, mean fluorescence intensity; SSC, side scatter.
Antibody and complement deposition is detectable on HOD × KEL RBC posttransfusion, with clearance of HOD × KEL RBC occurring in anti-KEL immunized recipients. (A) HOD × KEL RBC were labeled before transfusion into PBS (no antibody), anti-KEL–, or anti-HEL–treated WT mice. (B) IgG deposition was measured by direct antiglobulin test at 1, 2, and 24 hours posttransfusion. Total C3 (C) and active complement (C3b/iC3b) (D) was measured at 1, 2, and 24 hours posttransfusion. (E) RBC survival of HOD × KEL+ compared with HOD × KEL− RBC at various times posttransfusion. Means ± standard deviation (SD) shown. (A-D) ****P < .0001, ***P < .001, **P < .01, and not significant (ns) by 1-way ANOVA with Tukey multiple comparison test. (E) ****P < .0001 and ns by 2-way ANOVA with Dunnett multiple comparisons test. Data shown include 5 mice per group and are representative of 3 independent experiments. DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; DiO, 3,3′-dioctadecyloxacarbocyanine perchlorate; FSC, forward scatter; MFI, mean fluorescence intensity; SSC, side scatter.
Because antibodies bound to the surface of RBC have been shown to fix complement,40 we investigated whether complement was deposited on transfused HOD × KEL RBC. Complement deposition was determined by measuring total (C3) and active (C3b/iC3b) complement on transfused RBC at 1, 2, and 24 hours posttransfusion by flow cytometry. Complement deposition on transfused HOD × KEL RBC was significant only in recipient mice passively immunized with anti-KEL antibodies (Figure 2C-D). Specifically, total complement deposited on transfused cells in the anti-KEL–treated mice and increased over time (Figure 2C), peaking at 24 hours posttransfusion, whereas C3b/iC3b complement peaked at 1 hour (Figure 2D), indirectly indicating that complement on HOD × KEL RBC at 24 hours posttransfusion was residual bound complement product. These findings are consistent with previous reports using RBC expressing either the KEL or HOD antigens individually, where transfusion of KEL RBC in the presence of anti-KEL antibodies causes complement deposition but transfusion of HOD RBC in the presence of anti-HEL antibodies does not.40,44
Deposition of antibodies and complement on the RBC surface promote cell clearance from the circulation by opsonizing cells for removal.45-47 Because transfused HOD × KEL RBC were bound by antibody and, in the case of anti-KEL immunized mice, complement as well, we assessed RBC survival in all recipient groups. HOD × KEL RBC were not cleared in mice receiving anti-HEL antibody or PBS (Figure 2E). In contrast, in mice that received anti-KEL antibody, ∼20% of transfused HOD × KEL RBC were cleared. Importantly, RBC survival plateaued in this group and did not further significantly decrease between 4 and 24 hours posttransfusion (Figure 2E), which coincided kinetically with decreasing bound antibody (Figure 2B). These results suggest that noncleared HOD × KEL RBC persist by modification of antibody targets, such as antigen modulation, which has been shown to occur with single positive RBC.25 These results also demonstrate that the unique properties of each antigen are retained even when expressed on the same RBC, thus recapitulating findings when either antigen is expressed on RBC individually.40,44
Passive immunization results in antigen-specific antigen modulation
Antigen modulation has been shown to account for AMIS in models in which transfused RBC express a single foreign antigen and are not fully cleared from the circulation, likely related to decreased antibody binding on consequently antigen-negative cells.14,15 Because the majority of transfused HOD × KEL RBC survived in our model, despite antibody binding, we next assessed whether transfused HOD × KEL RBC underwent antigen loss. Antigen levels of KEL, HEL, and Ter119 were quantified on HOD × KEL RBC after transfusion into passively immunized recipients and compared with the levels of antigen detected in PBS control-treated recipients by flow cytometry. Whereas KEL antigen levels decreased on transfused HOD × KEL RBC over time in anti-KEL–treated mice, levels were unaffected in mice given anti-HEL antibodies or PBS (Figure 3A), indicating that passive immunization with anti-KEL antibody causes only KEL antigen loss, despite HOD and KEL antigens on the same RBC. Similarly, HEL antigen levels significantly decreased in mice that had received anti-HEL antibodies, but not in mice that had received anti-KEL antibodies or PBS control (Figure 3B), again indicating that antigen modulation on the RBC surface is antigen-specific. The decreases in KEL or HEL antigen levels on transfused HOD × KEL RBC in passively immunized recipients was not due to nonspecific membrane alterations of RBC because there was no difference among the groups in the levels of Ter119, a protein expressed on erythroid cells (Figure 3C).
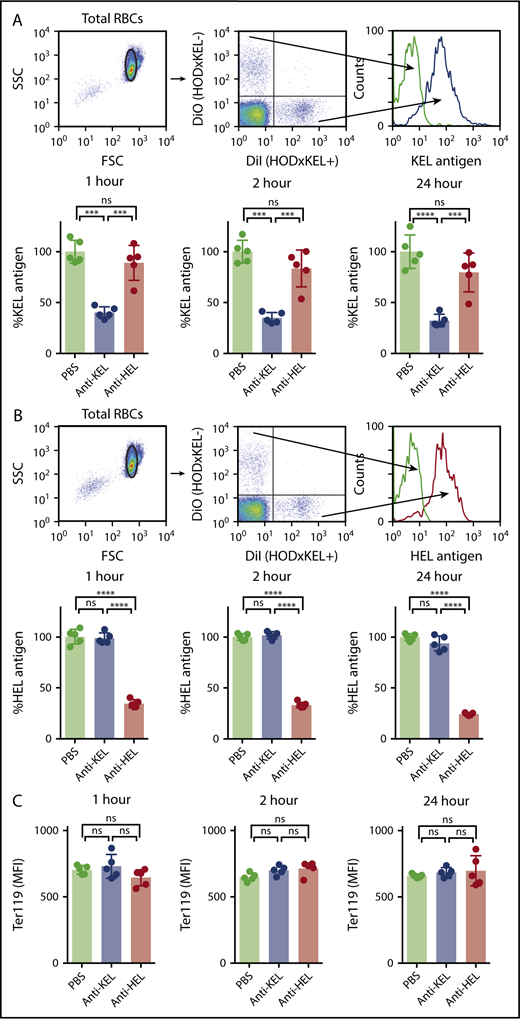
Antibody-induced decreases in the level of detectable antigen are antigen-specific. Posttransfusion, HOD × KEL RBC were stained for the level of detectable KEL antigen (A) or HEL antigen (B). (C) Transfused HOD × KEL RBC were stained for the level of detectable Ter119. Means ± SD shown. ****P < .0001, ***P < .001, and ns by 1-way ANOVA with Tukey multiple comparison test. Data shown include 5 mice per group and are representative of 3 independent experiments.
Antibody-induced decreases in the level of detectable antigen are antigen-specific. Posttransfusion, HOD × KEL RBC were stained for the level of detectable KEL antigen (A) or HEL antigen (B). (C) Transfused HOD × KEL RBC were stained for the level of detectable Ter119. Means ± SD shown. ****P < .0001, ***P < .001, and ns by 1-way ANOVA with Tukey multiple comparison test. Data shown include 5 mice per group and are representative of 3 independent experiments.
We next quantified the individual antigens on transfused RBC by western blot analysis to determine whether antibody-induced decreases in the level of detectable antigen truly reflected antigen-specific removal. WT mice were passively immunized with anti-KEL or anti-HEL antibodies, or given PBS control, and subsequently exposed to HOD × KEL RBC. Posttransfusion, the levels of KEL and HEL antigen were tracked over time until significant decreases occurred in the corresponding passively immunized mice. RBC were harvested to prepare RBC ghosts 48 hours posttransfusion, when maximal antigen loss of KEL (Figure 4A) and HEL antigens (Figure 4B), but not Ter119 (Figure 4C), was detected by flow cytometry, suggesting an optimal time point to facilitate detection of differences in antigen levels by western blot analysis. Consistent with the flow cytometry data, anti-KEL antibody induced antigen-specific removal of the KEL antigen, whereas anti-HEL antibody induced antigen-specific removal of the HEL antigen by western blot analysis (Figure 4D).

Antibody-induced decreases in the level of detectable antigen reflect antigen-specific removal and not RBC internalization. (A-C) Forty-eight hours posttransfusion into PBS, anti-KEL–, or anti-HEL–treated recipients, HOD × KEL RBC were stained for the level of detectable KEL antigen (A), HEL antigen (B), or Ter119 antigen (C). (D) HOD × KEL RBC ghosts or ghosts from nontransfused mice (NT) were analyzed by western blot by probing for KEL, HEL, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (E-G) HOD × KEL RBC were incubated with anti-KEL or anti-HEL antibodies in vitro at 4°C and 37°C for the indicated time and assessed for expression of KEL (E), HEL (F), and Ter119 (G) antigens by flow cytometry. Data in panels A-D and panels E-G are reflective of 2 independent experiments. ****P < .0001 and ns by 1-way ANOVA with Tukey multiple comparison test.
Antibody-induced decreases in the level of detectable antigen reflect antigen-specific removal and not RBC internalization. (A-C) Forty-eight hours posttransfusion into PBS, anti-KEL–, or anti-HEL–treated recipients, HOD × KEL RBC were stained for the level of detectable KEL antigen (A), HEL antigen (B), or Ter119 antigen (C). (D) HOD × KEL RBC ghosts or ghosts from nontransfused mice (NT) were analyzed by western blot by probing for KEL, HEL, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (E-G) HOD × KEL RBC were incubated with anti-KEL or anti-HEL antibodies in vitro at 4°C and 37°C for the indicated time and assessed for expression of KEL (E), HEL (F), and Ter119 (G) antigens by flow cytometry. Data in panels A-D and panels E-G are reflective of 2 independent experiments. ****P < .0001 and ns by 1-way ANOVA with Tukey multiple comparison test.
Although many cell types have dynamic cellular membranes, RBC are thought to lack the necessary machinery to endocytose surface antigens.48 However, we wanted to rule out the possibility that antibody-mediated internalization of antigen was responsible for the loss of detectable KEL and HEL antigens. HOD × KEL RBC were incubated with anti-KEL or anti-HEL antibodies in vitro at 37°C and assessed for expression of KEL or HEL antigens by flow cytometry at various time points. RBC incubated with anti-KEL antibody showed no difference in the level of KEL surface antigen when incubated at 37°C compared with 4°C at either 2 or 24 hours, nor was there any difference in the antigen level between the time points (Figure 4E). Similarly, RBC incubated with anti-HEL antibody showed no difference in HEL expression in various conditions and time points (Figure 4F). Ter119 antigen was likewise unaffected by anti-KEL or anti-HEL antibody treatment (Figure 4G). Thus, antigen loss on RBC does not appear to reflect antibody-mediated internalization of target antigen. Taken together, these results demonstrate that passive immunization results in removal of the KEL or HEL antigens from the RBC membrane without any detectable effect on other antigens.
Mechanism of antigen modulation is antigen-specific
We next explored potential mechanisms whereby KEL or HEL antigens were removed from transfused HOD × KEL RBC. Because macrophages are known to participate not only in the removal of whole RBC from circulation, but also pieces of RBC as well (eg, denatured hemoglobin, nuclear remnants), we hypothesized that antibody-binding to target antigen may result in antigen removal by phagocytic macrophages. To determine any role of macrophages in mediating antigen loss of antibody-bound transfused RBC, we conducted similar HOD × KEL RBC transfusion experiments in passively immunized WT mice following macrophage depletion by clodronate treatment, as done previously.35,36 Recipient mice were given empty (control) or clodronate liposomes via tail vein injection 2 days and 1 day before passive immunization and transfusion with HOD × KEL RBC (Figure 5A).
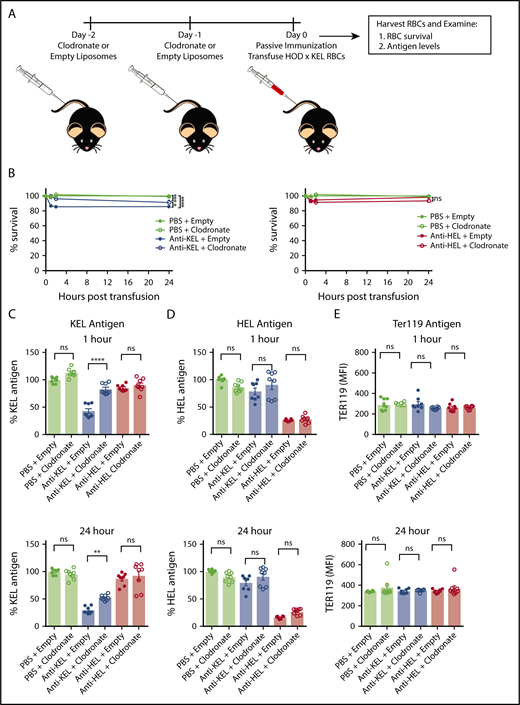
Macrophage-depletion abrogates KEL antigen loss in anti-KEL–treated recipients, but not HEL antigen loss in anti-HEL–treated recipients. (A) Recipients were treated with clodronate or empty (control) liposomes before passive immunization with either anti-KEL or anti-HEL antibodies, or PBS control, followed by transfusion of labeled HOD × KEL RBC. (B) HOD × KEL RBC survival at the indicated time points posttransfusion. Transfused HOD × KEL RBC were stained for the level of detectable KEL antigen (C), HEL antigen (D), or Ter119 antigen (E). Means ± SD shown. ****P < .0001, **P < .01, and ns by 2-way ANOVA with Dunnett multiple comparison test (B) and 1-way ANOVA with Tukey multiple comparison test (C-E). Data shown include 7 to 8 mice per group and are representative of 2 independent experiments.
Macrophage-depletion abrogates KEL antigen loss in anti-KEL–treated recipients, but not HEL antigen loss in anti-HEL–treated recipients. (A) Recipients were treated with clodronate or empty (control) liposomes before passive immunization with either anti-KEL or anti-HEL antibodies, or PBS control, followed by transfusion of labeled HOD × KEL RBC. (B) HOD × KEL RBC survival at the indicated time points posttransfusion. Transfused HOD × KEL RBC were stained for the level of detectable KEL antigen (C), HEL antigen (D), or Ter119 antigen (E). Means ± SD shown. ****P < .0001, **P < .01, and ns by 2-way ANOVA with Dunnett multiple comparison test (B) and 1-way ANOVA with Tukey multiple comparison test (C-E). Data shown include 7 to 8 mice per group and are representative of 2 independent experiments.
Survival of HOD × KEL RBC and antigen levels were assessed by flow cytometry at 1, 2, and 24 hours posttransfusion. Interestingly, macrophage depletion with clodronate significantly inhibited the clearance of KEL RBC in anti-KEL–treated recipients compared with empty liposome anti-KEL–treated recipients at all time points, although some decreased survival was observed even in clodronate-treated anti-KEL recipients at 24 hours compared with nonimmunized (PBS) control mice (Figure 5B). Anti-HEL–immunized recipients did not show evidence of HOD × KEL RBC clearance, which was unaffected by clodronate treatment.
Clodronate treatment did not significantly affect the level of KEL antigen on transfused cells in recipients given PBS control or immunized with anti-HEL antibody (Figure 5C). However, the extent of KEL antigen loss on transfused RBC at all time points in anti-KEL recipients was significantly inhibited when recipients were pretreated with clodronate, suggesting that modulation of the KEL antigen may be macrophage-dependent. In contrast, the decreased level of HEL antigen observed in anti-HEL–immunized recipients was unaffected by clodronate treatment (Figure 5D). Levels of Ter119 were not significantly different between recipients treated with empty vs clodronate liposomes (Figure 5E), once again suggesting that changes in antigen levels were antigen-specific.
The failure of clodronate-treatment to affect HEL antigen loss suggested involvement of an alternative mechanism of antigen modulation occurring independently of macrophages. For example, the HEL antigen may be uniquely sensitive to a mechanism that results in selective removal independent of phagocyte engagement, such as proteolytic cleavage. Therefore, we next explored potential differences between KEL and HEL antigens in terms of sensitivity to proteolytic cleavage by incubating HOD × KEL RBC with anti-KEL or anti-HEL antibodies and protease in vitro (Figure 6A). Although the KEL antigen failed to demonstrate sensitivity to protease treatment at any concentration tested (Figure 6B), the HEL antigen showed a dose-dependent effect of protease treatment (Figure 6C). Despite increased sensitivity of HEL to proteolytic cleavage, anti-HEL preincubation did not alter HEL removal in the presence of protease. Levels of the Ter119 antigen were unaffected by protease treatment (Figure 6D).

Protease treatment of HOD × KEL RBCs removes HEL but not KEL antigen. (A) HOD × KEL RBC were treated with various concentrations of protease following incubation with PBS control, anti-KEL, or anti-HEL antibodies in vitro. The level of detectable KEL antigen (B), HEL antigen (C), or Ter119 antigen (D) was determined by flow cytometry. Means ± SD shown. Data are reflective of 2 independent experiments.
Protease treatment of HOD × KEL RBCs removes HEL but not KEL antigen. (A) HOD × KEL RBC were treated with various concentrations of protease following incubation with PBS control, anti-KEL, or anti-HEL antibodies in vitro. The level of detectable KEL antigen (B), HEL antigen (C), or Ter119 antigen (D) was determined by flow cytometry. Means ± SD shown. Data are reflective of 2 independent experiments.
Suppression of antibody development (AMIS) is antigen-specific
Previous studies demonstrated a correlation between antigen modulation and suppression of sensitization in AMIS models in which RBC express a single foreign antigen.14,15 Therefore, we next tested recipient serum for newly generated anti-KEL and anti-HOD antibodies to determine whether antigen modulation correlated with AMIS and whether suppression was antigen-specific (Figure 7A). Anti-KEL IgM was detected on days 5 and 7 posttransfusion in mice that received PBS control or passive anti-HEL immunization (Figure 7B). However, anti-KEL IgM was not detected at any time points in mice that received anti-KEL immunization. Likewise, PBS control and anti-HEL–treated recipients developed anti-KEL IgG on days 7 and 14, but anti-KEL–treated recipients did not (Figure 7C). Thus, passive immunization with anti-KEL–protected recipients from developing endogenous anti-KEL antibodies following exposure to HOD × KEL RBC, whereas passive immunization with anti-HEL had no effect and resulted in an anti-KEL response similar to unimmunized PBS control-treated mice.
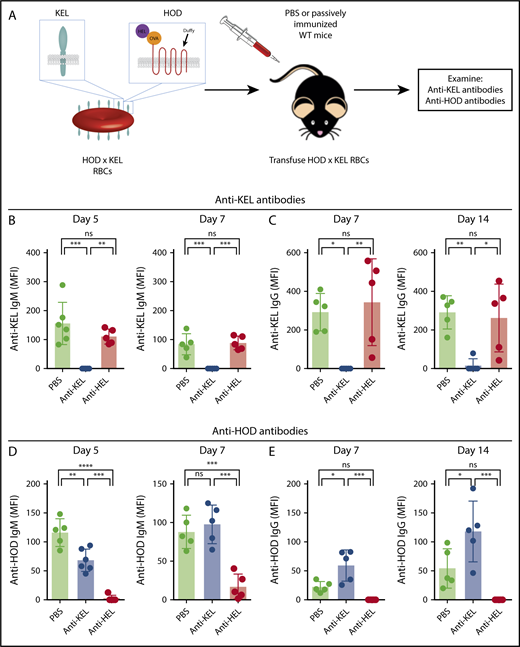
Exposure to antigen-specific antibodies induces antigen-specific antibody-mediated immunesuppression. WT mice were treated with PBS (no antibody), anti-KEL, or anti-HEL before exposure to HOD × KEL RBC (A) followed by evaluation of anti-KEL IgM (B), anti-KEL IgG antibodies (C), anti-HOD IgM (D), or anti-HOD IgG antibodies (E) at the time points indicated. Means ± SD shown. ****P < .0001, ***P < .001, **P < .01, *P < .05, and ns by 1-way ANOVA with Tukey multiple comparison test. Data shown include 5 mice per group and are representative of 3 independent experiments.
Exposure to antigen-specific antibodies induces antigen-specific antibody-mediated immunesuppression. WT mice were treated with PBS (no antibody), anti-KEL, or anti-HEL before exposure to HOD × KEL RBC (A) followed by evaluation of anti-KEL IgM (B), anti-KEL IgG antibodies (C), anti-HOD IgM (D), or anti-HOD IgG antibodies (E) at the time points indicated. Means ± SD shown. ****P < .0001, ***P < .001, **P < .01, *P < .05, and ns by 1-way ANOVA with Tukey multiple comparison test. Data shown include 5 mice per group and are representative of 3 independent experiments.
Determination of an anti-HOD response was similarly performed and yielded results consistent with the anti-KEL data. On days 5 and 7 posttransfusion, significant amounts of anti-HOD IgM was detected in both PBS control-treated and anti-KEL–treated mice, but not in mice that received passive anti-HEL immunization (Figure 7D). Additionally, anti-HOD IgG was detected at all time points in PBS control-treated and anti-KEL–treated mice, but not in mice that had received anti-HEL immunization (Figure 7E). Interestingly, anti-KEL immunized mice had a slightly, but significantly and reproducibly, increased anti-HOD IgG response compared with unimmunized PBS-control treated mice (Figure 7E), perhaps related to increased RBC clearance in anti-KEL–treated recipients (Figure 2E). Taken together, we find that mice passively immunized to KEL do not develop anti-KEL antibodies, but do develop anti-HOD antibodies after exposure to RBC bearing both antigens. Conversely, mice passively immunized to HEL do not develop anti-HOD antibodies, but do develop anti-KEL antibodies similarly to unimmunized mice, indicating that AMIS is antigen-specific.
Finally, we passively immunized recipients with both anti-KEL and anti-HEL antibodies and examined de novo antibody development (supplemental Figure 2A). Mice receiving dual antigen passive immunization experienced significantly reduced anti-KEL IgM (supplemental Figure 2B) and IgG (supplemental Figure 2C) antibodies and anti-HOD IgM (supplemental Figure 2D) and IgG (supplemental Figure 2E) antibodies, compared with nonimmunized PBS control-treated mice. These results are in agreement with the results in which recipients were immunized against a single antigen, because AMIS to a given antigen was observed only in the presence of passively administered antibody specific to that antigen.
Discussion
Here we report that passive immunization results in antigen-specific antigen modulation and suppression of antibody development. Using a novel AMIS model, in which mice were exposed to RBC bearing 2 foreign antigens and immunized with antibodies to either antigen individually, we tested the specificity of AMIS and any consequence on other nonspecific antigens. Murine RBC expressing both KEL and HOD RBC antigens demonstrated similar clearance kinetics, antigen levels, and antibody development after transfusion into passively immunized mice as is observed with KEL-only or HOD-only RBC. Furthermore, our data showed that antigen-specific immunization does not affect antibody binding, complement deposition, or antigen modulation to other RBC antigens.
Differences between the various animal models and clinical observations in humans have complicated our understanding of AMIS and how it occurs, but likely highlight the variability and multimechanistic nature of AMIS, depending on the particular antigens and antibodies involved. We intentionally used HOD × KEL RBC for our studies because antigen modulation, and not RBC clearance, which may cause nonspecific AMIS, appears to be the dominant mechanism in HOD-only or KEL-only models.14,15 Recently, in an AMIS model using HOD-only RBC and antiovalbumin antibodies, which resulted in both antigen modulation and RBC clearance, others reported that AMIS more closely correlates with saturating amounts of bound antibody and antigen modulation than with RBC clearance.14 Indeed, our data indicate that AMIS correlates more closely with IgG binding and antigen modulation than with RBC clearance. Even in the case of RhD, where RhIg results in significant RhD+ RBC clearance, we have previously found that RhIg induces antigen modulation in RhD+ patients that received RhIg as adjunct therapy for immune thrombocytopenic purpura.49 Thus, although RBC clearance occurs following antibody engagement with some antigens, the exact contribution of clearance in mediating AMIS requires further investigation.
Several prior studies implicate antigen modulation as a central pathway for inducing AMIS, which our current findings support, yet details regarding how antigen loss occurs have remained incompletely understood. Here we exclude antibody-mediated endocytosis of target antigen as the mechanism of RBC antigen loss, as well as identify a potential role for macrophages in mediating removal of the KEL, but not the HEL, antigen. However, because clodronate depletion may not be entirely specific to macrophages, our results do not rule out the potential role of other phagocytic populations, such as neutrophils, in antibody-mediated KEL antigen removal.50 Despite the differential effect of clodronate treatment on antigen modulation of KEL vs HEL, clodronate-treated recipients failed to generate de novo anti-KEL or anti-HOD antibodies regardless of antibody pretreatment (C.L.M. and S.R.S., unpublished observations). Thus, although clodronate treatment provided a mechanism to determine the potential role of macrophages and/or other phagocytes in antigen modulation following initial exposure to HOD × KEL RBC, this approach does not allow for direct examination of phagocyte-mediated antigen modulation on AMIS because these recipients fail to generate anti-KEL or anti-HEL antibodies even in the absence of antibody treatment. Future studies will be needed to examine the role of macrophage and/or other clodronate-sensitive cells in the development of alloantibodies following RBC transfusion.
Potential differences in mechanisms of antigen loss between KEL and HEL are also supported by our data demonstrating susceptibility of HEL, but not KEL, to protease treatment. Although HEL displayed sensitivity to proteolytic cleavage, anti-HEL antibodies failed to affect HEL antigen removal in this setting in vitro. One explanation for the lack of effect of antibody on protease sensitivity in vitro is that shear forces may be necessary for antibody-mediated antigen loss in vivo. Others have demonstrated shear-dependent proteolytic cleavage of cell surface proteins on platelets following ligand binding,51 and a similar mechanism involving endothelial or other cell-derived proteases could potentially affect HEL antigen loss from RBC. However, it is certainly possible that HEL antigen loss occurs through a protease-independent process that is likewise distinct from KEL. Nevertheless, these data suggest that macrophage-mediated removal of antibody-bound antigen may occur for certain RBC antigens, whereas other antigens may be lost through alternative means such as proteolytic degradation.
One notable but not completely surprising finding was the boosted anti-HOD IgG response in mice immunized with anti-KEL antibodies. A possible explanation for the enhanced response is that anti-KEL–related RBC clearance creates a more inflammatory microenvironment. Inflammation is known to contribute to RBC alloimmunization because it can favorably increase anti-HOD antibody formation following HOD-only RBC transfusion.2,52,53 Similar findings have been reported following transfusion of KEL RBC into recipients pretreated with the inflammation-inducing agent, poly (I:C).43,53,54 Poly (I:C) not only enhances anti-KEL antibodies following KEL RBC transfusion,43,53,54 but also directly increases anti-HOD antibody formation following subsequent HOD × KEL RBC transfusion.43 In contrast, no boost in anti-KEL IgG occurred in mice immunized with anti-HEL antibody in our present study, compared with PBS control-treated mice, perhaps because anti-HEL did not induce RBC clearance and associated inflammatory signals.
Although differences in the consequences of antibody engagement of antigen may contribute to the distinct immunological outcomes observed following HOD × KEL RBC transfusion in the presence of anti-KEL or anti-HEL antibodies, unique characteristics of the antigens themselves might also influence these varied responses. Recent reports demonstrate that antigen density can affect antibody development, with lower density antigen inducing a state of antigen-specific nonresponsiveness.33 It is certainly possible that antigen density after initial antigen loss may be variable and influence the likelihood of subsequent antibody formation. Differences in the effect of anti-KEL and anti-HEL may also reflect differences in the antibodies used; polyclonal anti-KEL antibody preparation was collected from immunized donors, whereas anti-HEL antibodies were simply a mixture of 2 purified monoclonal antibodies. Our findings demonstrate that the developing immune response and AMIS may vary depending on the combination of passively administered antibody and RBC antigen(s) involved. Whether differences in antibodies used and/or distinct characteristics of the antigens themselves dictated unique outcomes following HOD × KEL RBC transfusion remains to be determined.
Understanding AMIS will allow for the development or improved understanding of various efficacious antibodies with clinical utility. This includes production of monoclonal antibodies to prevent RBC alloimmunization to a range of RBC alloantigens, inhibiting sensitization to non-RBC antigens by monoclonal or polyclonal agents, and even understanding how currently available antibody-based therapeutics affect RBC. For example, recently we found that antigen modulation occurs on human RBC after treatment with daratumumab, an anti-CD38 monoclonal antibody used in the treatment of multiple myeloma.42 Furthermore, intravenous immunoglobulin is known to cause antibody deposition on RBC and, in some cases, hemolytic anemia.55-58 Such findings highlight the importance of considering off-target effects of additional antibody-based therapeutic interventions.
As with all experimental models, limitations of using mice to recapitulate the pathophysiology of human disease must be considered. We and others consider the use of HOD or KEL RBC in AMIS studies to be more physiologically relevant, especially compared with models using transfusion of sheep RBC into mice.14,20 As an advantage, our study models AMIS using 2 antigens that contain clinically relevant human proteins; however, allogeneic RBC transfused to human patients express a panoply of foreign antigens, each of which may elicit unique mechanisms of AMIS in the presence of antibody. Such mechanistic multiplicity likely contributes to whether AMIS is antigen-specific, again depending on the particular antibodies and antigens involved. Whether antigen modulation occurs as a common mechanism of AMIS to additional antigens, and in humans, requires additional investigation.
In conclusion, we observed antigen-specific AMIS secondary to antigen modulation for both the HOD and KEL antigens in our model, indicating that the immune response following antigen-specific immunization does not affect antibody binding, antigen modulation, or antibody development to other RBC antigens. We also provide evidence suggesting a role for macrophages in mediating antigen loss of the KEL, but not the HEL, antigen, which highlights the distinct properties of different antigens. Our study provides insight into the specificity of AMIS resulting from antigen modulation, which may enlighten the development of additional therapeutics based on this type of immune suppression.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Emory University Integrated Cellular Imaging Microscopy Core, especially Laura Fox-Goharioon for assistance with super resolution microscopy.
This work was supported by grants from the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) (R01 HL138656, R01 HL13557, and P01 HL132819) (S.R.S.), NIH Director’s Early Independence Award Program (DP5OD019892) (S.R.S.), and the Burroughs Wellcome Trust Career Award to Medical Scientists (S.R.S.); NIH, NHLBI training grant (T32HL069769) (C.L.M.); and NIH, NHLBI predoctoral fellowship (1F31HL131428-01) (A.M.).
Authorship
Contribution: C.L.M., A.M., S.R.P., R.P.J., A.L.B., and C.M.A. performed the experiments; C.L.M. and A.M. wrote the initial manuscript, which was commented on and edited by all authors; and C.L.M., A.M., S.R.P., J.E.H., and S.R.S. oversaw the experimental design.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sean R. Stowell, Center for Transfusion and Cellular Therapies, Department of Laboratory Medicine and Pathology, Emory University School of Medicine, 615 Michael St, Atlanta, GA 30322; e-mail: srstowe@emory.edu.

