

Key Points
Hemophilia A carriers had a significantly reduced and less sustained FVIII response to DDAVP compared with control patients.
Hemophilia A carriers with higher bleeding scores had significantly lower DDAVP responses.

Abstract
The cause of hemophilia A carrier bleeding is not well established. Desmopressin (DDAVP), used clinically to treat or prevent bleeding, can also be used as a medical stress surrogate. This study’s objective was to compare the response to DDAVP in hemophilia A carriers with that in normal control patients. Bleeding was assessed by the International Society on Thrombosis and Hemostasis Bleeding Assessment Tool (ISTH-BAT). DDAVP (0.3 μg/kg) was administered either IV or subcutaneously (SC), and blood was drawn at baseline and 1, 2, and 4 hours postadministration. Blood was assessed for factor VIII (FVIII) level, von Willebrand factor (VWF) antigen (VWF:Ag), VWF activity (VWF:RCo or VWF:GPIbM), thromboelastography (TEG), and thrombin generation assay (TGA) at all points, and for VWF propeptide (VWFpp):Ag ratio and ABO blood type at baseline. Carriers were older than control patients (median age, 34 and 21 years, respectively; P = .003) and had higher ISTH-BAT bleeding scores (median bleeding score, 8 and 0, respectively; P = .001). Carriers had a significantly reduced FVIII response to DDAVP compared with control patients (P ≤ .0001). When only carriers with normal baseline FVIII levels (n = 10) were included, carriers maintained a reduced FVIII response (P ≤ .0001). Furthermore, participants with abnormal bleeding scores had a significantly lower FVIII response to DDAVP compared with those with normal bleeding scores (P = .036). Hemophilia A carriers have a lower and less sustained FVIII response to DDAVP, suggesting an impaired ability to respond to hemostatic stress, which contributes to bleeding.
Introduction
Hemophilia A is a rare bleeding disorder caused by a deficiency in factor VIII (FVIII) affecting approximately 1 in every 5000 males.1 Because of the X-linked inheritance pattern of the disease, males are affected with hemophilia, whereas females are carriers. Historically, female carriers were assumed to be asymptomatic; however, it is now known that many hemophilia carriers experience abnormal bleeding symptoms.2
Although the normal range of circulating FVIII in the general population is 0.5 to 1.5 IU/mL, hemophilia carriers present with a wide range of FVIII levels. About 30% of hemophilia carriers have low FVIII levels and can have abnormal bleeding symptoms.2 However, recent studies demonstrate that even hemophilia A carriers with normal factor levels can have abnormal bleeding phenotypes.2,3 In fact, FVIII level has been found to be weakly correlated with bleeding scores in carriers.3,4 Accordingly, it has been suggested that circulating factor level is not a good predictor of bleeding tendency in hemophilia carriers.4
It is important to better understand the cause of hemophilia carrier bleeding, as it can have a major effect on health and quality of life. Hemophilia A carriers primarily experience excessive mucocutaneous bleeding; notably, heavy menstrual bleeding and postpartum bleeding.2,3 Excessive bleeding after surgery, tooth extraction, and trauma is also common. However, hemarthrosis has been reported and is supported by bleeding score and magnetic resonance imaging evidence.3,5
Desmopressin (DDAVP) is a synthetic analog of vasopressin. DDAVP increases plasma VWF, FVIII, and tissue plasminogen activator (t-PA); causes vasodilation; and increases platelet procoagulant activity.6-9 DDAVP has been used for decades to treat mild hemophilia A, von Willebrand disease (VWD), and platelet disorders. Nevertheless, the cellular mechanisms of the hemostatic effects of the drug remain incompletely characterized. The mechanism of action of DDAVP-induced VWF increase is generally accepted to be DDAVP binding directly to vasopressin 2 receptors on endothelial cells, resulting in the release of VWF from Weibel Palade bodies.10,11 However, the mechanism of action of FVIII increase remains poorly understood. Two primary theories exist. The first suggests that DDAVP induces FVIII release directly from its producing cells.12,13 Conversely, the second proposes that FVIII increases are indirect and that FVIII is protected from degradation through DDAVP-induced increases in VWF.14
Because of its hemostatic effect, DDAVP can be used as a medical stress surrogate to mimic in vivo hemostatic response to vessel injury. Using this feature, this study aims to understand the mechanism of abnormal bleeding in hemophilia A carriers by comparing the laboratory response to DDAVP in hemophilia A carriers and normal control patients.
Methods
Research subjects
Carriers of hemophilia A, at least 18 years of age, were eligible for this study. Recruitment occurred through the Inherited Bleeding Disorders Clinics in Kingston, ON, Canada, and Atlanta, GA. Hemophilia A carriers either had a known F8 mutation or an appropriate family history (the daughter of a man with hemophilia A or the mother of 2 sons with hemophilia A or the mother of 1 son with hemophilia A plus 1 other affected male relative). Carriers were excluded if they were pregnant or using hormonal therapy or contraceptives. All subjects provided informed consent. Ethics approval was obtained from the Queen’s University Health Sciences and Affiliated Teaching Hospitals Research Ethics Board and the Emory University Institutional Review Board.
Normal control patients were recruited in Kingston through several methods, including the Queen’s Paid Research Studies Facebook page, the Kingston General Hospital Research Institute website, flyers posted in Kingston General Hospital, and an advertisement in the Kingston newspaper (The Kingston Whig-Standard). Normal control patients were healthy women aged 18 to 50 years. Potential control patients were excluded if they had any significant medical history; smoked; had a history of vascular disease, myocardial infarction, stroke, or venous thromboembolism; were using hormonal therapy, hormonal contraceptives, or corticosteroids; were pregnant; or were unable to follow the 24-hour fluid restriction protocol. All subjects provided informed consent.
Data collection
All subjects had their bleeding assessed by the International Society on Thrombosis and Hemostasis Bleeding Assessment Tool (ISTH-BAT). Blood was taken before DDAVP administration. Subjects were then administered 0.3 μg/kg DDAVP (maximum 20 μg), either IV or subcutaneously (SC). Blood was drawn at 1, 2, and 4 hours postadministration. FVIII:C, VWF:Ag, and VWF activity (VWF:GPIbM assay in Kingston and VWF:RCo in Atlanta) were assessed at all points. ABO blood type for all subjects and F8 genotype in hemophilia A carriers was determined or documented if available from previous studies. In addition, in Kingston, thromboelastography (TEG), thrombin generation assay (TGA), and a FVIII chromogenic assay were evaluated at all points, and VWFpp was measured at baseline.
Laboratory analysis
VWF:Ag, VWF activity (VWF:RCo or VWF:GPIbM) and FVIII:C
VWF:Ag, VWF:GPIbM, and FVIII:C were assessed in Kingston in the Kingston General Hospital core laboratory. Assays at Emory University were performed in the coagulation laboratory, with the only difference in protocol being the use of VWF:RCo assay for VWF activity. The VWF:RCo assay was performed using a Siemens kit on a Siemens BCS XP system.
FVIII chromogenic assay
The FVIII chromogenic assay was performed using a Siemen’s kit and the manufacturer’s assay conditions. The assay was performed on the Siemens Sysmex CA 1500. Patient plasma samples, as well as normal control patient and pathogenic control patient plasma, were evaluated at 1/1 and 1/2 dilutions.
VWFpp
VWFpp was measured using the Immucor VWF and propeptide assay kit (303292). The assay was performed according to the manufacturer’s specifications. The plate was read in a fluorescent plate reader with excitation wavelengths between 315 and 340 nm and emission wavelengths between 370 and 470 nm.
ABO blood type
DNA extraction was performed using the Qiagen Gentra Puregene kit according to the manufacturer’s specifications. Extracted DNA was hydrated with DNA hydration solution (Qiagen kit) in microcentrifuge tubes and stored at −80°C.
ABO blood type was determined by polymerase chain reaction, as described by Lee et al.15
Thromboelastography
Whole blood was drawn into 3.2% sodium citrate, and was assayed within 2 hours. All subjects had phlebotomy performed by the same nurse, using a tourniquet, and after resting for several minutes. TEG was performed using the TEG 5000 Thromboelastograph Hemostasis Analyzer System (Haemonetics). Next, 340 μL whole blood was added to 20 μL of 0.2 M CaCl2 in the reaction cup without activator (native), and a TEG tracing was recorded.
Thrombin generation
TGA was performed using Diapharma Technothrombin TGA reagents according to the manufacturer’s recommendations. Briefly, a calibration curve was established using the thrombin calibrator (Diapharma). Subsequently, TGA reagent C low (RC low, Diapharma), patient plasma, and chromogenic substrate were added to a black NUNC 96-well maxisorp plate and read kinetically in a fluorescent plate reader at 360/460 nm for 1.5 hours.
F8 genotype
F8 mutation severity was classified as severe and nonsevere on the basis of the FVIII gene (F8) variant database (http://www.factorviii-db.org). This database compiles cases of variants in the F8 gene and currently reports 2015 unique variants for hemophilia A. The database reports, on the basis of these cases, whether a mutation produces mild, moderate, or severe disease. For this study, mutations causing mild and moderate disease in males (per the F8 variant database) were grouped as nonsevere mutations, whereas mutations causing severe disease in males were classified as severe mutations.
Statistical analysis
All statistical analysis was performed using Graphpad Prism 6. Comparisons of hemophilia A carrier and normal control patient values were performed using Mann-Whitney U tests. Two-way repeated measures analysis of variance with Bonferonni multiple comparisons was used to analyze the time course response to DDAVP in hemophilia A carriers and normal control patients. Pearson correlation was performed using age and the peak change in FVIII to assess whether FVIII response to DDAVP decreases with age.
Results
Subjects and baseline levels
Seventeen hemophilia A carriers and 9 normal control patients were enrolled in the study. Because of sample clotting, 2 normal control patients were excluded from all analysis. As shown in Table 1, Hemophilia A carriers were significantly older than normal control patients, with median ages of 34 and 21 years, respectively (P = .003). As expected, hemophilia A carriers had significantly higher ISTH-BAT bleeding scores (BS) compared with normal control patients (medians of 8 and 0, respectively; P = .001). The distribution of O-type blood did not differ between the 2 groups, with 53% of hemophilia A carriers and 57% of normal control patients having type O blood (P = .872). Median baseline FVIII levels were significantly lower in hemophilia A carriers compared with normal control patients (0.55 and 1.20 IU/mL, respectively; P = .001). Median baseline VWF:Ag and VWF activity did not differ between hemophilia A carriers and control patients (0.77 vs 0.82 IU/mL [P = .067] and 0.55 vs 0.67 [P = .266], respectively). The median VWFpp:Ag ratio in hemophilia A carriers was 1.10, whereas in control patients it was significantly higher, at 1.50 (P = .047).
Baseline levels for hemophilia A carriers and control patients
| Hemophilia A carriers (n=17) | Normal controls (n = 7) | |
|---|---|---|
| Median age (range), y | 34 (25-60) | 21* (18-45) |
| Median ISTH-BAT BS (range) | 8 (0-18) | 0* (0-4) |
| Blood type O, % | 53 (n = 13) | 57 (n = 7) |
| Median FVIII level (range), IU/mL | 0.55 (0.31-1.25) | 1.20* (0.82-1.98) |
| Median VWF:Ag (range), IU/mL | 0.77 (0.32-1.68) | 0.82 (0.55-1.35) |
| Median VWF activity (range), IU/mL† | 0.55 (0.35-1.66) | 0.67 (0.48-1.44) |
| Median VWFpp:Ag ratio (range) | 1.10 (0.6-1.8) (n = 8) | 1.50‡ (1.2-1.9) (n = 5) |
| Hemophilia A carriers (n=17) | Normal controls (n = 7) | |
|---|---|---|
| Median age (range), y | 34 (25-60) | 21* (18-45) |
| Median ISTH-BAT BS (range) | 8 (0-18) | 0* (0-4) |
| Blood type O, % | 53 (n = 13) | 57 (n = 7) |
| Median FVIII level (range), IU/mL | 0.55 (0.31-1.25) | 1.20* (0.82-1.98) |
| Median VWF:Ag (range), IU/mL | 0.77 (0.32-1.68) | 0.82 (0.55-1.35) |
| Median VWF activity (range), IU/mL† | 0.55 (0.35-1.66) | 0.67 (0.48-1.44) |
| Median VWFpp:Ag ratio (range) | 1.10 (0.6-1.8) (n = 8) | 1.50‡ (1.2-1.9) (n = 5) |
P ≤ .01.
VWF:GPIbM or VWF:RCo.
P ≤ .05.
FVIII response to DDAVP
Analysis of FVIII response to DDAVP showed that hemophilia A carriers had a significantly reduced overall response to DDAVP compared with control patients (P ≤ .0001; Figure 1). In addition, although hemophilia A carriers had significantly lower FVIII levels before the administration of DDAVP (P ≤ .05), multiple comparison analysis revealed that the disparity in FVIII level was amplified in response to DDAVP treatment, with carriers having significantly lower FVIII levels at the 1-, 2-, and 4-hour points (all P ≤ .0001). The slope of the FVIII response (defined as [peak FVIII level − pre FVIII level]/time to peak) was also less steep in hemophilia A carriers than normal control patients (0.72 and 1.20, respectively; P = .004).

FVIII response to DDAVP in hemophilia A carriers and control patients. Hemophilia A carriers (red) and normal control patients (blue). *P ≤ .05. ****P ≤ .0001.
FVIII response to DDAVP in hemophilia A carriers and control patients. Hemophilia A carriers (red) and normal control patients (blue). *P ≤ .05. ****P ≤ .0001.
Subsequent analysis to determine whether the reduced FVIII response to DDAVP was maintained in hemophilia A carriers with normal baseline FVIII levels (n = 10; median baseline FVIII = 0.71 IU/mL) revealed that carriers with normal baseline FVIII levels had a significantly reduced overall FVIII response to DDAVP compared with control patients (P ≤ .0001; Figure 2). Importantly, multiple comparison analysis demonstrated that the difference in FVIII response was not significant at baseline, but was significant at the 1-, 2-, and 4-hour points (1-, 2-, and 4-hour all P ≤ .0001).
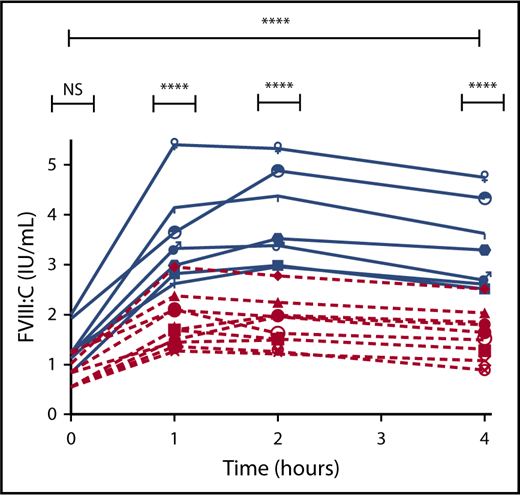
FVIII response to DDAVP in hemophilia A carriers with normal baseline FVIII levels and control patients. Hemophilia A carriers with normal baseline FVIII levels (red) and normal control patients (blue). ****P ≤ .0001. NS, not significant.
FVIII response to DDAVP in hemophilia A carriers with normal baseline FVIII levels and control patients. Hemophilia A carriers with normal baseline FVIII levels (red) and normal control patients (blue). ****P ≤ .0001. NS, not significant.
FVIII chromogenic vs 1-stage assay
A FVIII chromogenic assay was performed and compared with the FVIII 1-stage assay. Hemophilia A carriers (for whom a plasma sample was available, n = 7) and normal control patients (n = 7) were included, and the results from the 2 assays were compared at all 4 points, giving a total of 56 results on which to compare the 2 assays. There was a very strong Pearson correlation of 0.895 between the 1-stage assay and the chromogenic assay (P = .002).
VWF:Ag and VWF activity
VWF:Ag response to DDAVP did not differ between hemophilia A carriers and control patients (P = .559; Figure 3). Similarly, VWF activity (VWF:RCo or VWF:GPIbM) response to DDAVP treatment was not significantly different between carriers and control patients (P = .171; Figure 3).

VWF responses to DDAVP in hemophilia A carriers and control patients. VWF:Ag (A) and VWF activity (VWF:RCo or VWF:GPIbM) (B) response to DDAVP in carriers (red) and control patients (blue). P = NS.
VWF responses to DDAVP in hemophilia A carriers and control patients. VWF:Ag (A) and VWF activity (VWF:RCo or VWF:GPIbM) (B) response to DDAVP in carriers (red) and control patients (blue). P = NS.
VWF:RCo vs VWF:GPIbM
VWF:RCo and VWF:GPIbM data were available for 6 hemophilia A carriers because of their inclusion in a previous study. The values obtained using the 2 VWF activity assays were compared. There was a very strong Pearson correlation of 0.900 between the VWF:RCo and the VWF:GPIbM assays (P = .015).
FVIII/VWF:Ag ratio
The FVIII/VWF:Ag ratio was assessed both pre-DDAVP administration and at peak. Hemophilia A carriers had significantly lower FVIII/VWF:Ag ratio than control patients at both points (0.66 vs 1.51 [P ≤ .0001] and 0.92 vs 2.22 [P ≤ .0001]). FVIII/VWF:Ag ratio was also assessed for a correlation with bleeding score in our subjects (both carriers and control patients); however, no statistically significant correlation was found.
DDAVP response in hemophilia A carriers with severe and nonsevere mutations
DDAVP response was assessed in hemophilia A carriers with severe mutations and compared with carriers with nonsevere mutations. Fifteen of the 17 hemophilia A carriers were included in the analysis (1 carrier was excluded because the mutation was described in severe, moderate, and mild hemophilia, and 1 carrier was excluded because the mutation was not documented in the F8 variant database). F8 mutations are shown in Table 2. DDAVP responses were not significantly different between the 2 groups for FVIII (shown in Figure 4), VWF:Ag, or VWF activity (P = .230, .720, and .422, respectively).
F8 mutations in hemophilia A carriers
| Mutation, exon | Domain | Type | Mutation severity | N | FVIII levels |
|---|---|---|---|---|---|
| p.(Pro151Ser), 4 | A1 | Missense | Mild | 2 | 0.57, 0.84 |
| p.(Tyr365Cys), 8 | A1 | Missense | Mild | 1 | 1.21 |
| Intron 22 | Inversion | Severe | 5 | 0.31, 0.83, 0.35, 0.58, 0.57 | |
| p.(Arg1800His), 16 | A3 | Missense | Mild/moderate/severe | 1 | 1.03 |
| p.(Leu1193Glnfs*27), 14 | B | Dup. fs. | Severe | 2 | 0.55, 0.42 |
| p.(Arg2159Cys), 23 | C1 | Missense | Mild | 1 | 0.54 |
| p.(Asn1941Ser), 18 | A3 | Missense | Severe | 1 | 0.42 |
| p.(Gly720Arg), 14 | Missense | 1 | 0.35 | ||
| p.(Arg2169Cys), 23 | C1 | Missense | Mild | 1 | 0.35 |
| p.(Arg602*), 12 | A2 | Nonsense | Severe | 1 | 1.25 |
| p.(Ser554Gly), 11 | A2 | Missense | Mild | 1 | 0.39 |
| Mutation, exon | Domain | Type | Mutation severity | N | FVIII levels |
|---|---|---|---|---|---|
| p.(Pro151Ser), 4 | A1 | Missense | Mild | 2 | 0.57, 0.84 |
| p.(Tyr365Cys), 8 | A1 | Missense | Mild | 1 | 1.21 |
| Intron 22 | Inversion | Severe | 5 | 0.31, 0.83, 0.35, 0.58, 0.57 | |
| p.(Arg1800His), 16 | A3 | Missense | Mild/moderate/severe | 1 | 1.03 |
| p.(Leu1193Glnfs*27), 14 | B | Dup. fs. | Severe | 2 | 0.55, 0.42 |
| p.(Arg2159Cys), 23 | C1 | Missense | Mild | 1 | 0.54 |
| p.(Asn1941Ser), 18 | A3 | Missense | Severe | 1 | 0.42 |
| p.(Gly720Arg), 14 | Missense | 1 | 0.35 | ||
| p.(Arg2169Cys), 23 | C1 | Missense | Mild | 1 | 0.35 |
| p.(Arg602*), 12 | A2 | Nonsense | Severe | 1 | 1.25 |
| p.(Ser554Gly), 11 | A2 | Missense | Mild | 1 | 0.39 |
Dup. fs., duplication frameshift.
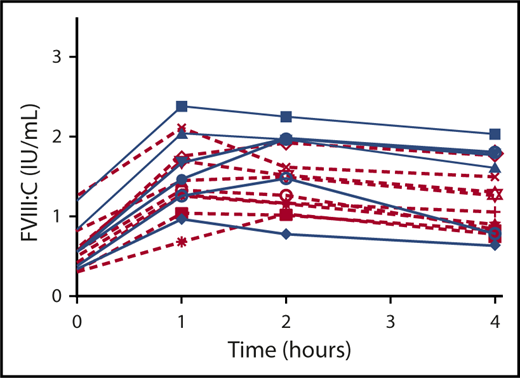
FVIII response to DDAVP in hemophilia A carriers with severe and nonsevere mutations. Severe mutations (blue) and nonsevere mutations (red). P = NS.
FVIII response to DDAVP in hemophilia A carriers with severe and nonsevere mutations. Severe mutations (blue) and nonsevere mutations (red). P = NS.
FVIII response to DDAVP and age
Pearson correlation was calculated using all study subjects (carriers and control patients) to assess whether FVIII response to DDAVP decreases with age; however, the results were not significant (r = −0.33; P = .12).
Thrombin generation and thromboelastography
A TGA was performed on plasma samples from hemophilia A carriers (for whom a plasma sample was available, n = 7) and normal control patients (n = 7) at all points and assessed for lag time (minutes), peak thrombin (nM), peak time (minutes), velocity index, and area under the curve. There were no significant differences between carriers and control patients in lag time, peak time, velocity index, or area under the curve for any times. Peak thrombin was not significantly different between carriers and control patients for the pre, 1-hour, and 2-hour times. However, median peak thrombin was significantly different at the 4-hour point (P = .038). The median peak thrombin at 4 hours in hemophilia A carriers was 441.6 nM, whereas in control patients, it was 287.1 nM.
TEG was performed for carriers (for whom a whole blood sample was available, n = 7) and control patients (n = 7) for the pre and 1-hour times and assessed for R (time), K (time), α-angle (degrees), and maximum amplitude (MA, in mm). None of the criteria evaluated were significantly different between carriers and control patients at either point.
ISTH-BAT and FVIII
FVIII response to DDAVP comparing subjects (both hemophilia A carriers and normal control patients) who had normal ISTH-BAT BS with subjects who had abnormal ISTH-BAT BS showed that subjects with abnormal BS had a significantly lower FVIII response to DDAVP compared with subjects with normal BS (P = .036; Figure 5).
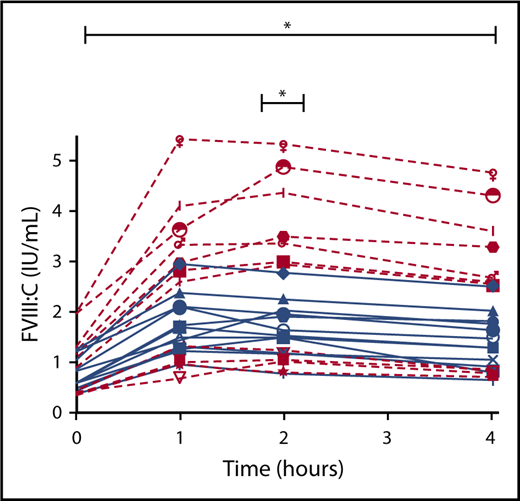
FVIII response to DDAVP in subjects with normal BS (<6) and abnormal BS (≥6). Normal BS (red) and abnormal BS (blue). *P ≤ .05.
FVIII response to DDAVP in subjects with normal BS (<6) and abnormal BS (≥6). Normal BS (red) and abnormal BS (blue). *P ≤ .05.
ISTH-BAT BS in carriers
Carriers’ ISTH-BATs were examined to determine which components were high for most carriers. We found that menstrual bleeding was the primary issue, with a mean ISTH-BAT BS of 2.24. In addition, tooth extraction bleeding and cutaneous bleeding were the second and third highest mean scores in carriers (1.35 and 1.06, respectively). The mean postpartum BS in carriers was 0.47.
Discussion
Although it is recognized that carriers of hemophilia experience abnormal bleeding, the underlying cause of this bleeding has not been well established. This is especially significant, given that residual plasma FVIII level has been found to be an ineffective predictor of hemophilia carrier bleeding in contrast to its disease defining accuracy in males. Using DDAVP to mimic in vivo hemostatic response to vessel injury, this study aimed to explore the mechanism of bleeding in hemophilia carriers.
We revealed that hemophilia A carriers have a reduced ability to elevate and sustain FVIII levels in response to DDAVP. Importantly, analysis of hemophilia A carriers with normal baseline FVIII levels showed that the difference in FVIII response between carriers and control patients was not a result of baseline FVIII level but, rather, FVIII response to DDAVP. Furthermore, this study demonstrated that study participants with elevated ISTH-BAT BS had significantly reduced FVIII responses to DDAVP compared with those with normal ISTH-BAT BS. These findings provide a possible explanation for baseline FVIII level as a poor indicator of carrier bleeding and suggest it may be the FVIII response to hemostatic injury that is critical. This inability to increase and sustain FVIII levels may explain why hemophilia carriers often experience excessive and prolonged bleeding in response to trauma or injury. Baseline circulating FVIII levels may be sufficient to prevent spontaneous bleeds in most carriers; however, their inability to respond sufficiently to hemostatic stress may result in prolonged and excessive bleeding.
Interestingly, these results suggest that a F8 mutation may be implicated in more than just reducing the circulating level of FVIII. A F8 mutation may also alter the storage and release of FVIII from cells in response to hemostatic stress. Further investigation is warranted as to the effect of a F8 mutation on FVIII synthesis, storage, and release, and in particular in the cellular stored pools of FVIII.
Hemophilia A carriers had similar VWF:Ag and VWF activity (VWF:RCo or VWF:GPIbM) responses to DDAVP compared with control patients. This is consistent with previous findings of Kobrinsky et al16 and Casonato et al,17 who compared the FVIII and VWF:Ag responses with DDAVP in hemophilia A carriers and normal control patients as a way of improving carrier detection before genetic testing.16,17 Both groups found a reduced increase in FVIII, but normal increases in VWF:Ag in response to DDAVP in hemophilia A carriers compared with healthy females. This suggests that VWF response to hemostatic stress is not impaired in carriers.
The finding that FVIII responses to DDAVP differed between carriers and control patients, but that VWF:Ag and VWF:RCo responses did not, suggests that FVIII increase in response to DDAVP is not mediated by VWF but, rather, that DDAVP acts directly to cause release of FVIII from cells. This supports the theory of an in vivo cellular storage pool of FVIII that releases FVIII in response to hemostatic stress and signaling. This indicates that there may be a cell type that expresses vasopressin 2 receptors and can store and release FVIII that is not currently known, as it has been suggested that liver sinusoidal endothelial cells do not express vasopressin 2 receptors.18 Furthermore, these results indicate that the conventional relationship between FVIII and VWF, that FVIII levels are dependent on VWF levels, may be incomplete. FVIII levels may be able to increase separately from and in addition to the increases caused by increased levels of circulating VWF. However, because of the uncertainty of the mechanism of DDAVP-mediated FVIII increase and of FVIII release in vivo, it is not currently possible to determine the cause of the reduced FVIII response to DDAVP shown in hemophilia A carriers in this study. Further investigation as to the mechanism of DDAVP-induced FVIII increase and of FVIII release in vivo are necessary.
Although genotype is a strong predictor of disease severity in males, the literature is mixed as to the influence of F8 genotype on bleeding in hemophilia A carriers.19-21 Unlike males with hemophilia A, female carriers have 1 normal F8 in addition to their mutated copy. The normal F8 allele may mask the influence of mutation severity in carriers, or conversely, the mutated allele may be deleterious to the production of FVIII from the normal allele to varying degrees. Further exploration as to the consequences of a F8 in hemophilia A carriers is warranted.
With respect to the global assays of hemostasis, TGA and TEG, the only significant finding was a higher 4-hour peak thrombin in carriers. We attempted to see whether there were any distinguishing features in those carriers with higher 4-hour peak thrombin. We found that the 2 carriers with the lowest 4-hour peak thrombin (270.2 and 273.2) had the highest BS (17 and 18). However, 4 normal control patients also had 4-hour peak thrombin values in the 200s (range, 219.5-287.1), but all had normal BS. The carriers (n = 5) with the highest 4-hour peak thrombin (range 437.1-490.7) had BS ranging from 4 to 13 and baseline FVIII ranging from 0.3 to 0.9. Thus, it is difficult to ascertain any specific characteristic that may distinguish these carriers from those with lower 4-hour peak thrombin values.
It is interesting that all the parameters evaluated for TGA, other than 4-hour peak thrombin discussed here, showed no differences between carriers and control patients, given that there was a significant reduction in FVIII response to DDAVP in the hemophilia A carriers, and that the use of TGA in male patients with hemophilia has been found to correlate with FVIII activity and bleeding symptom phenotype.22,23 However, TGA may be less useful when FVIII:C is closer to normal, as in hemophilia carriers. As such, comparing FVIII:C values that are closer to normal to normal FVIII:C may be difficult. In addition, TGA is a global assay of hemostasis, and as such, it is possible that other factors are masking the reduced FVIII response. For instance, varying amounts of tissue factor trigger have been shown to modulate the extent to which the intrinsic pathway contributes to thrombin generation.24,25 In addition, TGA does not entirely mimic in vivo conditions. Because platelet-poor plasma is used, no platelets are present during the assay. It might be useful to attempt TGA using whole-blood samples to better mimic in vivo hemostasis; however, in our study, TEG analysis using whole blood also did not reveal any differences between carriers and control patients. This was unexpected, as TEG in male patients with hemophilia has also been found to be correlated with FVIII activity and clinical bleeding pattern, and to be useful for monitoring bypass agent therapy in this population.22,26,27 Similar to TGA, TEG is a global assay of hemostasis, and as such, other factors may be masking the reduced FVIII response to DDAVP in carriers.
Interestingly, subjects with abnormal ISTH-BAT BS had reduced FVIII responses to DDAVP. However, there were 4 subjects who had very reduced FVIII responses but normal BS. All 4 of these subjects were carriers of hemophilia A. The ages of these carriers were 25, 29, 29, and 37 years. Because of their relatively young age, they may not have had sufficient hemostatic challenge to accumulate bleeding events. However, all had experienced hemostatic challenge in the form of pregnancy/birth, tooth extraction, or trauma/surgery, with 3 of these carriers having experienced 2 of the 3 challenges listed here, whereas 1 had experienced all 3 challenges. ISTH-BAT menstrual BS were 0, 1, 1, and 3. Total BS were 0, 1, 3, and 4. Alternatively, hemophilia carrier bleeding may be multifactorial, and other factors may be contributing to their tendency toward a normal bleeding phenotype.
DDAVP was administered IV in Atlanta, and primarily SC in Kingston, because of differences in usual clinical practice. All control patients received SC DDAVP, whereas 9 carriers received SC DDAVP and 8 carriers received IV DDAVP. Although SC DDAVP has been previously found to be similar to IV DDAVP, differences have been reported by some groups. De Sio et al found that the response to SC and IV DDAVP was not different, whereas Mannucci et al found that IV DDAVP caused a shorter time to peak and a higher peak DDAVP level compared with SC.28,29 Given these previous findings, the differences in routes of administration may have had an effect on the results.
There are several limitations to this study in addition to the differences in administration of DDAVP discussed here. The significant age difference between carriers and control patients may have influenced our results, as both VWF and FVIII levels have been shown to be modulated by age.30 Carriers recruited in this study were older than control patients, and given that BS increase with age, this could have influenced the finding of carriers having higher BS than control patients.31 In addition, only subjects recruited in Kingston were able to have certain assays performed, reducing the sample size for these assays. Timing of menstrual periods was not controlled for in participants. The literature is mixed as to whether hemostatic factors vary throughout the menstrual cycle. A systemic review by Knol et al in 2012 reported 5/11 studies included in their analysis showed cyclic variation in VWF:Ag and 2/9 studies showed variation in FVIII:C.32 As such, it is difficult to discern the influence of not controlling for menstrual cycle on our results. Finally, our data do not allow us to determine whether any older subjects had perimenopausal bleeding, as the ISTH-BAT only quantifies whether heavy menstrual bleeding started at menarche or not.
This study has improved the understanding of the cause of abnormal bleeding in hemophilia A carriers, suggesting that a contributor to this bleeding phenotype may be an inability to generate and sustain a sufficient increase in FVIII in response to hemostatic stress. This may be a result of the F8 mutation impairing FVIII synthesis, storage, and/or release. An improved understanding of the mechanism of hemophilia A carriers bleeding may make it possible to accurately assess bleeding risk and provide better healthcare to these women.
Acknowledgment
This study was supported in part by research funding from the Hemostasis and Thrombosis Research Society Mentored Research Award.
Authorship
Contribution: V.C. contributed to the design of the study, performed experiments, collected data, analyzed and interpreted the data, and drafted the manuscript; P.D.J. designed the study, supervised the research, analyzed and interpreted the data, and drafted the manuscript; R.F.S. designed the study, recruited subjects, and collected data; H.W. recruited subjects and collected data; W.M.H. assisted with statistical analysis; D.G. coordinated sample assessment; L.T. performed phlebotomy and performed the desmopressin trials; J.G. recruited subjects and collected data; M.B. contributed to experiments; L.H. performed thromboelastography; and all authors revised the manuscript.
Conflict-of-interest disclosure: P.D.J. has received research funding from CSL Behring, Bayer, and Shire. R.F.S. has received research funding from Bioverativ, Kedrion/Grifols, Genentech, and Shire and has received honoraria from Shire, Genentech/Roche, CSL Behring, Bioverativ, Uniqure, Biomarin, Bayer, and Novo Nordisk. The remaining authors declare no competing financial interests.
Correspondence: Paula D. James, Department of Medicine, Queen's University, Room 2015 Etherington Hall, 94 Stuart St, Kingston ON K7L 3N6, Canada; e-mail: jamesp@queensu.ca.

