

Key Points
Steroid-refractory GI GVHD is a risk factor for complement-mediated TA-TMA, driven by alternative and terminal pathway activation.
Concomitant TA-TMA and GI GVHD is associated with abysmal OS, longer hospital stays, and greater health care costs.

Abstract
Transplant-associated thrombotic microangiopathy (TA-TMA), a complication of hematopoietic cell transplant (HCT), is associated with significant morbidity and mortality. The pathophysiology and overlap of TA-TMA with other posttransplant complications such as graft-versus-host disease (GVHD) is poorly understood. We retrospectively identified cases of TA-TMA among patients with grade 3/4 gastrointestinal (GI) GVHD, reviewed intestinal biopsy specimens, and performed correlative testing of biomarkers associated with TA-TMA. TA-TMA was more common in patients with steroid-refractory GVHD compared with steroid-responsive GVHD (79.3% vs 42.1%; P = .001). Among patients surviving 100 days post-HCT, 1-year survival from day 100 was significantly better for patients who had not developed TA-TMA in the first 100 days (69.5% vs 36.7%; P < .001). Only 1 of 7 proposed TA-TMA histology criteria (mucosal hemorrhage) differed significantly based on GVHD steroid response. In multivariable modeling, steroid-refractory GVHD was a risk factor for development of TA-TMA (hazard ratio, 3.09; 95% confidence interval, 1.68-5.67; P < .001). There were no differences in complement activation at GVHD onset; however, 2 to 6 weeks later, patients with TA-TMA had higher levels of BBPlus and C5b-9, markers of alternative and terminal pathway activation (BBPlus: median, 600 vs 209.3 ng/mL; P = .0045) (C5b-9: median, 425.9 vs 258.4 ng/mL; P = .029). TA-TMA is associated with poor overall survival (OS) following HCT and may be detected early by histologic findings and may be differentiated from GVHD by measurement of alternative and terminal complement pathway activation. It is unknown whether treatment of TA-TMA will improve survival in steroid-refractory GVHD.
Introduction
Transplant-associated thrombotic microangiopathy (TA-TMA) is an increasingly recognized complication of hematopoietic cell transplant (HCT) and is associated with significant morbidity and mortality.1 Early descriptions of the incidence of TA-TMA were based largely on retrospective autopsy studies and ranged from 40% to 46% with reported nonrelapse mortality (NRM) rates of 30% to 45%.2-4 TA-TMA causes systemic endothelial injury and can affect the central nervous system, cardiovascular system, lungs, kidney, connective tissue, and gastrointestinal (GI) tract, similar to other types of TMA such as atypical hemolytic uremic syndrome (aHUS) and thrombotic thrombocytopenic purpura (TTP).1 Consensus clinical diagnostic criteria have been published by both the Blood and Marrow Transplantation Clinical Trials Network (BMT CTN) and by the International Working Group (IWG).5,6 An increased risk of bleeding or lack of bona-fide target organ often prevents biopsy, thereby precluding histological findings as diagnostic criteria. In validation studies, about one-fifth of patients met IWG criteria but not BMT CTN criteria due to lack of renal or neurologic manifestations. More than two-thirds of patients met BMT CTN criteria but not IWG due to schistocytosis less than the cutoff value of 4% or 8 per high power field.7,8
Despite increased recognition as a post-HCT complication, TA-TMA has remained a significant cause of NRM because of ineffective treatment options.9-11 Complement activation has been reported in other TMAs including aHUS and acquired TTP.12-14 Based on this evidence, the complement inhibitor, eculizumab, was used to treat plasma exchange–resistant cases of aHUS leading to its approval by the US Food and Drug Administration in 2013.14 In pediatric TA-TMA, an elevated level of C5b-9, a marker of terminal complement activation, has been associated with poor prognosis.15 Based on experience with aHUS and evidence of complement activation in TA-TMA, a number of pediatric and adult TA-TMA patients have been successfully treated with eculizumab.16-19 Evidence of complement activation and response to complement inhibition have led to the theory that endothelial damage in the post-HCT setting leads to complement activation, an important driver of TA-TMA.
Acute graft-versus-host disease (GVHD) is another major cause of NRM.20-23 First-line treatment with steroids achieves durable remission in no more than 50%, and second-line therapies are associated with high failure rates and poor overall survival (OS).24 Immune-modulating therapies targeting cytokine receptors have been explored, though none have demonstrated superior efficacy and there remains no standard of care for second-line treatment of acute steroid-refractory GVHD.25-27 The pathophysiology of acute GVHD can be partially attributed to donor T-cell activation, but the lack of an effective second-line immunosuppressive therapy suggests that our understanding of GVHD pathophysiology is incomplete or that other processes may lead to steroid-refractoriness. A growing body of literature supports the role of endothelial dysfunction in the pathophysiology of acute GVHD.28-31 Luft and colleagues have demonstrated an association between steroid-refractoriness and renal TA-TMA and provide evidence of increased markers of endothelial injury in patients with renal TA-TMA.32 We believe that complement activation is a downstream effect of endothelial injury and, as such, may serve as a mechanistic link between GVHD and TA-TMA.33-35
In this study, we hypothesized that the proportion of patients with TA-TMA would be higher among those with steroid-refractory GVHD compared with those with steroid-responsive GVHD. We also hypothesized that patients with steroid-refractory GVHD of the GI tract would be more likely to have transfusion-dependent thrombocytopenia and/or anemia, and would demonstrate histologic evidence of TA-TMA. To evaluate our hypotheses, we reviewed intestinal biopsy specimens from patients with GI GVHD for histological findings consistent with TA-TMA. To investigate a link between steroid-refractory GVHD and TA-TMA via endothelial dysfunction and resultant complement activation, we measured plasma biomarkers of the classical, alternative, and terminal complement pathways.
Methods
Patients
We studied patients who underwent allogeneic HCT and were admitted to The Ohio State University Comprehensive Cancer Center between October 2008 and October 2016. Follow-up was complete through March 2018. Under a protocol approved by the institutional review board, we identified 124 patients who had been diagnosed with grade 3/4 GI GVHD and had undergone at least 1 endoscopy with biopsy after onset of GVHD symptoms. TA-TMA diagnosis was made retrospectively based on criteria encompassing cytopenias, hemolysis, and renal dysfunction. The specific TA-TMA diagnostic criteria and GVHD steroid-response criteria are defined in Table 1. Patients were considered to have TA-TMA if they met at least 2 of 3 diagnostic criteria. Clinical and laboratory data were abstracted from the electronic medical record. Lactate dehydrogenase (LDH) was documented as elevated, normal, or missing with elevation defined by 2 consecutive measurements greater than the upper limit of normal (ULN) occurring at least 30 days post-HCT. End points of interest included hospital readmissions, length of stay (LOS), and OS.
Definitions of TA-TMA criteria and GVHD steroid responses
| Definition | |
|---|---|
| TMA criteria | |
| Anemia/thrombocytopenia | Received >2 transfusions of platelets and/or packed red blood cells after initial GI biopsy |
| Hemolysis | Documentation of schistocytes or nucleated red blood cells on at least 2 consecutive tests |
| Renal end-organ damage | Evidence of new or worsening hypertension based on documentation or addition/titration of medication, acute kidney injury based on Acute Kidney Injury Network criteria, or proteinuria of ≥30 mg/dL by urinalysis, spot protein/creatinine ratio, or 24-hour urine collection |
| GVHD steroid responses | |
| Steroid-refractory | Lack of clinical response after 7 days of at least 2 mg/kg per day of methylprednisolone or equivalent dose |
| Steroid-dependent | Requirement for a second course of at least 1 mg/kg per day after an attempt to taper |
| Steroid-responsive | Any case not meeting criteria for steroid-refractory or steroid-dependent |
| Definition | |
|---|---|
| TMA criteria | |
| Anemia/thrombocytopenia | Received >2 transfusions of platelets and/or packed red blood cells after initial GI biopsy |
| Hemolysis | Documentation of schistocytes or nucleated red blood cells on at least 2 consecutive tests |
| Renal end-organ damage | Evidence of new or worsening hypertension based on documentation or addition/titration of medication, acute kidney injury based on Acute Kidney Injury Network criteria, or proteinuria of ≥30 mg/dL by urinalysis, spot protein/creatinine ratio, or 24-hour urine collection |
| GVHD steroid responses | |
| Steroid-refractory | Lack of clinical response after 7 days of at least 2 mg/kg per day of methylprednisolone or equivalent dose |
| Steroid-dependent | Requirement for a second course of at least 1 mg/kg per day after an attempt to taper |
| Steroid-responsive | Any case not meeting criteria for steroid-refractory or steroid-dependent |
Histopathological diagnosis
All cases accessioned for each patient after the time of GVHD symptom onset, as determined by chart review, were reviewed. Some patients underwent endoscopic biopsy on multiple occasions during post-HCT care yielding a total of 237 cases for review. All specimens were fixed in formalin and embedded in paraffin. Sections were stained with hematoxylin-eosin. Results of immunohistochemical staining for infections were noted when present. Specimens were reviewed by an expert pathologist (M.Y.) in a blinded manner. Eight signs of TA-TMA were previously proposed and studied by El-Bietar et al.36 These 8 signs and others associated with GVHD or infectious colitis were documented as present or absent.
Plasma complement markers
Fifty of the 124 patients in our study were enrolled in at least 1 prospective GVHD biomarker study for which specimens were collected and stored. Complement biomarkers were assessed at 2 time points: in the week before first intestinal biopsy and again 2 to 6 weeks later depending on sample availability. Per institutional standard, most patients underwent endoscopy at first presentation of GI GVHD symptoms so these time points were selected to approximate GI GVHD onset. In total, 47 patients had complement biomarker measurements from at least 1 time point of interest. Plasma was used in preference to serum based on previously published work demonstrating better sample stability over time for frozen specimens.37
Statistical analysis
Medians, ranges, frequencies, and percentages were used to describe patient characteristics. The Wilcoxon (Mann-Whitney U) rank-sum test was used to compare transfusion requirement between GVHD steroid response groups, and to compare complement marker levels at GVHD onset, 2 to 6 weeks post-GVHD, and changes from GVHD onset to 2 to 6 weeks later between TA-TMA and no TA-TMA groups. The Fisher exact test was used to compare histology findings among GVHD steroid response groups. Time to TA-TMA was defined as days from transplantation. Univariable and multivariable Fine and Gray regression analyses were preformed to evaluate the association between potential risk factors and TA-TMA, treating death prior to TA-TMA as a competing risk. OS was defined from the date of transplantation to the date of death, censoring those alive at the last follow-up date. Cox proportional hazard models were used to evaluate the association with risk of death. Risk factors considered for modeling include patient age, disease, disease status, Hematopoietic Cell Transplantation Comorbidity Index score, pre-HCT creatinine, HLA match, sex match, ABO match, stem cell source, stem cell dose, preparative regimen, and use of antithymocyte globulin (ATG). GVHD onset, GVHD steroid response, and onset of bacterial, viral, cytomegalovirus (CMV), or fungal infections were treated as time-dependent covariates in the model. Risk factors with significance level of P < .20 from univariable analyses were further evaluated in a multivariable analysis using a stepwise selection procedure, retaining those with P < .05 in the final model. Landmark analysis was used to evaluate the association between TA-TMA status by day 100 posttransplantation and OS. Kaplan-Meier curve was generated and compared using the log-rank test. The significance level was set at 0.05 and no adjustment was made for multiple testing. Statistics software Stata 14 was used for all analyses.
Results
Patient characteristics
The median age at transplant was 54 years (range, 19-72 years) and 40.3% were female. Reduced-intensity or nonmyeloablative conditioning was used in 66.1% of patients and all but 11 patients received a tacrolimus-containing GVHD prophylaxis regimen. The median time to GVHD onset was 38 days (range, 12-273 days). The prevalence of steroid-refractory, -dependent, and -responsive GVHD was 46.8%, 22.6%, and 29.8%, respectively. Additional patient characteristic data can be found in supplemental Table 1. The overall incidence of TA-TMA in the study population was 67.7%. Among 84 patients with both GVHD and TA-TMA, TA-TMA predated GVHD onset in only 8 patients. The median time to onset of TA-TMA post-HCT was 83.5 days (range, 17-2448 days). TA-TMA was diagnosed after GVHD onset in 77.4% of steroid-refractory GVHD patients, 76.0% of steroid-dependent GVHD patients, and 42.1% of steroid-responsive GVHD patients. The differences in TA-TMA prevalence between steroid-refractory and -responsive and steroid-dependent and -responsive GVHD patients were statistically significant (P = .001 and .01, respectively). LDH was elevated in 83.3% of patients with TA-TMA and only 52.5% of patients without TA-TMA (P = .001). LDH data were missing for 10.7% of TA-TMA patients and 27.5% of patients without TA-TMA.
Higher transfusion requirements in patients with steroid-refractory GVHD
In the 4 weeks after GVHD onset, patients with steroid-refractory GVHD required more platelet and packed red blood cell (pRBC) transfusions than patients with steroid-dependent or -responsive GVHD (Figure 1). Differences in platelet and pRBC transfusion requirements were noted as early as 2 weeks after GVHD onset between steroid-refractory and -responsive patients (P = .038 and P = .0085, respectively). Liberal transfusion parameters are often used for patients with GI bleeding. To account for potential confounding of the diagnosis of TA-TMA, subgroup analysis of patients with GI bleeding was performed. Of the 24 cases of chart-documented GI bleeding, the majority occurred in steroid-refractory patients (n = 14). The incidence of GI bleeding was highest in steroid-dependent GVHD patients, followed by steroid-refractory and steroid-responsive (26%, 24%, and 8%, respectively; P = .07). In patients with GI bleeding, those with TA-TMA required more platelet (median, 23 vs 12; P = .045) and pRBC transfusions (median, 19 vs 7; P = .02) than those without. In patients without documented GI bleeding, those with TA-TMA required more pRBC transfusions (median, 9 vs 3; P = .03), but not platelet transfusions (median, 5 vs 3; P = .21), than those without TA-TMA.
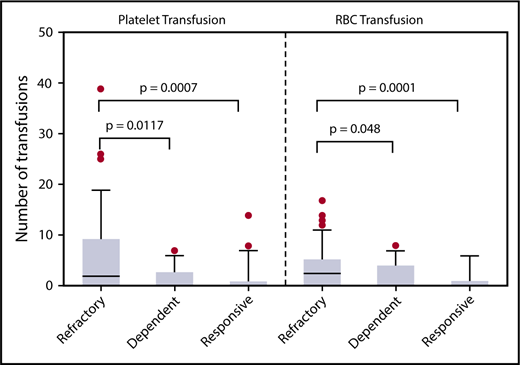
Patients with steroid-refractory GVHD required more transfusions than patients with steroid-dependent or steroid-responsive GVHD. Platelet and pRBC transfusions administered in the first 4 weeks after endoscopy for GI GVHD symptoms are shown. Steroid-refractory GVHD patients required more platelets and pRBCs than either steroid-dependent or steroid-responsive GVHD patients. The increased transfusion requirement supports our hypothesis that steroid-refractory GVHD patients may have concurrent TA-TMA explaining their lack of response to increasing immune suppression.
Patients with steroid-refractory GVHD required more transfusions than patients with steroid-dependent or steroid-responsive GVHD. Platelet and pRBC transfusions administered in the first 4 weeks after endoscopy for GI GVHD symptoms are shown. Steroid-refractory GVHD patients required more platelets and pRBCs than either steroid-dependent or steroid-responsive GVHD patients. The increased transfusion requirement supports our hypothesis that steroid-refractory GVHD patients may have concurrent TA-TMA explaining their lack of response to increasing immune suppression.
Histologic characteristics associated with TA-TMA
The patterns of histologic findings within 7 days of GVHD onset, presented by GVHD steroid response, are found in Table 2. Among the previously proposed TMA-associated histology findings, there were significant or marginally significant associations with GVHD steroid response. Between-groups comparisons revealed some marginally significant associations, specifically, mucosal hemorrhage was present in 100% of steroid-refractory cases and only 83.3% of steroid-dependent cases (P = .07), endothelial cell swelling was present in 62.5% of steroid-refractory cases compared with 36.4% of steroid-responsive cases (P = .10), and loss of glands was noted in 59.4% of steroid-refractory cases compared with 31.8% of steroid-responsive cases (P = .06). There were no significant differences between groups for any of the histologic findings associated with viral infection. There was a significant difference in depth of epithelial cell apoptosis with deeper levels of involvement for patients with steroid-refractory GVHD compared with steroid-responsive GVHD. Full-depth involvement was seen in 34.4% of steroid-refractory cases compared with 0% of steroid-responsive cases (P = .01). There was also a greater prevalence of mucosal erosion in steroid-refractory cases compared with steroid-responsive (43.8% vs 13.6%, P = .04). Photomicrographs depicting select TA-TMA and GVHD characteristics can be found in supplemental Figure 1.
Presence of histology findings by GVHD steroid response
| Histology finding | Steroid-refractory GVHD, n = 32 | Steroid-dependent GVHD, n = 12 | Steroid-responsive GVHD, n = 22 | P | |||||
|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | Overall | Refractory vs responsive | Refractory vs dependent | |
| TMA-associated findings | |||||||||
| Mucosal hemorrhage | 32 | 100.0 | 10 | 83.3 | 21 | 95.5 | .04 | .41 | .07 |
| Loss of glands | 19 | 59.4 | 5 | 41.7 | 7 | 31.8 | .13 | .06 | .33 |
| Intraluminal schistocytes | 2 | 6.3 | 0 | 0.0 | 0 | 0.0 | .67 | .51 | .99 |
| Intraluminal fibrin/microthrombi | 4 | 12.5 | 2 | 16.7 | 4 | 18.2 | .82 | .70 | .66 |
| Endothelial cell swelling | 20 | 62.5 | 4 | 33.3 | 8 | 36.4 | .10 | .10 | .10 |
| Endothelial cell separation | 5 | 15.6 | 1 | 8.3 | 1 | 4.5 | .59 | .38 | .99 |
| Areas of totally denuded mucosa | 17 | 53.1 | 4 | 33.3 | 7 | 31.8 | .26 | .17 | .32 |
| GVHD-associated findings | |||||||||
| Mucosal erosion | 14 | 43.8 | 2 | 16.7 | 3 | 13.6 | .04 | .04 | .16 |
| Lamina propria inflammatory infiltrate | 7 | 21.9 | 2 | 16.7 | 2 | 9.1 | .46 | .28 | .99 |
| Cystic gland dilation | 16 | 50.0 | 4 | 33.3 | 8 | 36.4 | .57 | .41 | .50 |
| Apoptotic crypt abscesses | 20 | 62.5 | 5 | 41.7 | 12 | 54.5 | .49 | .59 | .31 |
| Neutrophilic crypt abscesses | 10 | 31.3 | 1 | 8.3 | 6 | 27.3 | .32 | .99 | .24 |
| Acute inflammation of lamina propria | 15 | 46.9 | 6 | 50.0 | 9 | 40.9 | .85 | .78 | .99 |
| Ulceration | 7 | 21.9 | 3 | 25.0 | 2 | 9.1 | .37 | .28 | .99 |
| Depth of epithelial cell apoptosis (crypt/gland) | .01 | .01 | .37 | ||||||
| One-third depth | 14 | 43.8 | 8 | 66.7 | 12 | 54.6 | |||
| Two-thirds depth | 6 | 18.8 | 1 | 8.3 | 9 | 40.9 | |||
| Full depth | 11 | 34.4 | 2 | 16.7 | 0 | 0.0 | |||
| None | 1 | 3.1 | 1 | 8.3 | 1 | 4.6 | |||
| Infection-associated findings | |||||||||
| CMV inclusion bodies or +IHC stain | 0 | 0.0 | 1 | 8.3 | 2 | 9.1 | .18 | .16 | .27 |
| Adenovirus inclusion bodies or +IHC stain | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | |||
| Histology finding | Steroid-refractory GVHD, n = 32 | Steroid-dependent GVHD, n = 12 | Steroid-responsive GVHD, n = 22 | P | |||||
|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | Overall | Refractory vs responsive | Refractory vs dependent | |
| TMA-associated findings | |||||||||
| Mucosal hemorrhage | 32 | 100.0 | 10 | 83.3 | 21 | 95.5 | .04 | .41 | .07 |
| Loss of glands | 19 | 59.4 | 5 | 41.7 | 7 | 31.8 | .13 | .06 | .33 |
| Intraluminal schistocytes | 2 | 6.3 | 0 | 0.0 | 0 | 0.0 | .67 | .51 | .99 |
| Intraluminal fibrin/microthrombi | 4 | 12.5 | 2 | 16.7 | 4 | 18.2 | .82 | .70 | .66 |
| Endothelial cell swelling | 20 | 62.5 | 4 | 33.3 | 8 | 36.4 | .10 | .10 | .10 |
| Endothelial cell separation | 5 | 15.6 | 1 | 8.3 | 1 | 4.5 | .59 | .38 | .99 |
| Areas of totally denuded mucosa | 17 | 53.1 | 4 | 33.3 | 7 | 31.8 | .26 | .17 | .32 |
| GVHD-associated findings | |||||||||
| Mucosal erosion | 14 | 43.8 | 2 | 16.7 | 3 | 13.6 | .04 | .04 | .16 |
| Lamina propria inflammatory infiltrate | 7 | 21.9 | 2 | 16.7 | 2 | 9.1 | .46 | .28 | .99 |
| Cystic gland dilation | 16 | 50.0 | 4 | 33.3 | 8 | 36.4 | .57 | .41 | .50 |
| Apoptotic crypt abscesses | 20 | 62.5 | 5 | 41.7 | 12 | 54.5 | .49 | .59 | .31 |
| Neutrophilic crypt abscesses | 10 | 31.3 | 1 | 8.3 | 6 | 27.3 | .32 | .99 | .24 |
| Acute inflammation of lamina propria | 15 | 46.9 | 6 | 50.0 | 9 | 40.9 | .85 | .78 | .99 |
| Ulceration | 7 | 21.9 | 3 | 25.0 | 2 | 9.1 | .37 | .28 | .99 |
| Depth of epithelial cell apoptosis (crypt/gland) | .01 | .01 | .37 | ||||||
| One-third depth | 14 | 43.8 | 8 | 66.7 | 12 | 54.6 | |||
| Two-thirds depth | 6 | 18.8 | 1 | 8.3 | 9 | 40.9 | |||
| Full depth | 11 | 34.4 | 2 | 16.7 | 0 | 0.0 | |||
| None | 1 | 3.1 | 1 | 8.3 | 1 | 4.6 | |||
| Infection-associated findings | |||||||||
| CMV inclusion bodies or +IHC stain | 0 | 0.0 | 1 | 8.3 | 2 | 9.1 | .18 | .16 | .27 |
| Adenovirus inclusion bodies or +IHC stain | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | |||
IHC, immunhistochemistry.
Risk factors for development of TA-TMA
The multivariable Fine and Gray regression model of the risk of developing TA-TMA is shown in Table 3. Compared with patients with steroid-responsive GVHD, patients with steroid-refractory GVHD were more likely to develop TA-TMA (hazard ratio [HR], 3.09; 95% confidence interval [CI], 1.68-5.67; P < .001). The same was true for steroid-dependent GVHD patients compared with steroid-responsive patients (HR, 2.42; 95% CI, 1.17-4.98; P = .017). Viral infections other than CMV and fungal infections, occurring prior to TA-TMA onset, were also associated with increased risk of developing TA-TMA. The use of ATG in conditioning regimen and having not yet developed GVHD were protective factors. None of the other considered factors were associated with risk of TA-TMA including conditioning regimen, HLA match, stem cell source, and GVHD prophylaxis.
Multivariable Fine and Gray model of risk of developing TA-TMA
| HR | 95% CI | P | ||
|---|---|---|---|---|
| ATG use | 0.47 | 0.30 | 0.75 | .001 |
| GVHD status* | ||||
| Not yet developed | 0.35 | 0.14 | 0.88 | .025 |
| Steroid-responsive | 1.00 | |||
| Steroid-refractory | 3.09 | 1.68 | 5.67 | <.001 |
| Steroid-dependent | 2.42 | 1.17 | 4.98 | .017 |
| Viral infection other than CMV | 2.15 | 1.37 | 3.38 | .001 |
| Fungal infection | 5.87 | 1.35 | 25.55 | .018 |
| HR | 95% CI | P | ||
|---|---|---|---|---|
| ATG use | 0.47 | 0.30 | 0.75 | .001 |
| GVHD status* | ||||
| Not yet developed | 0.35 | 0.14 | 0.88 | .025 |
| Steroid-responsive | 1.00 | |||
| Steroid-refractory | 3.09 | 1.68 | 5.67 | <.001 |
| Steroid-dependent | 2.42 | 1.17 | 4.98 | .017 |
| Viral infection other than CMV | 2.15 | 1.37 | 3.38 | .001 |
| Fungal infection | 5.87 | 1.35 | 25.55 | .018 |
GVHD status was treated as a time-dependent covariate in the model.
Markers of complement activation in TA-TMA
At GVHD onset, there were no significant differences in plasma levels of any complement marker between patients with and without TA-TMA (Table 4). Interestingly, median C4d levels (classical pathway) for both groups were higher than the ULN for our laboratory. Median levels of C5a and C5b-9 (terminal pathway) were at or around the ULN whereas median BBPlus levels (alternative pathway) were well within normal limits. At follow-up sampling 2 to 6 weeks later, patients with TA-TMA demonstrated significantly higher levels of BBPlus (alternative pathway) and C5b-9 (terminal pathway) levels (Table 4). The change in complement over time was evaluated in patients with and without TA-TMA. A significant difference in the change in BBPlus (alternative pathway) from GVHD onset to 2 to 6 weeks later was noted. Specifically, BBPlus levels at GVHD onset were similar in patients with and without TA-TMA but decreased over time only in patients without TA-TMA (Figure 2).
Complement markers by TA-TMA status at GVHD onset and 2 to 6 weeks later
| Markers (complement pathway) | Normal values, ng/dL | TA-TMA + GVHD | GVHD only | |||
|---|---|---|---|---|---|---|
| n | Median (range) | n | Median (range) | P | ||
| GVHD onset | ||||||
| BBPlus (alternative) | 244-961 | 21 | 403.2 (124.5-2323.0) | 14 | 481.5 (239.8-1816.6) | .87 |
| C5b-9 (terminal) | 34-248 | 21 | 289.8 (147.5-643.0) | 14 | 263.7 (169.1-518.3) | .46 |
| C4d (classical) | 279-1846 | 21 | 2854.8 (962.0-5016.2) | 14 | 2678.6 (1399.7-6205.7) | .74 |
| C5a (terminal) | 19-48 | 21 | 52.0 (13.2-105.2) | 14 | 42.5 (18.8-89.1) | .71 |
| 2-6 wk after GVHD onset | ||||||
| BBPlus (alternative) | 244-961 | 29 | 600.0 (130.6-2119.0) | 12 | 209.3 (94.5-908.7) | .0045 |
| C5b-9 (terminal) | 34-248 | 29 | 425.9 (193.2-860.0) | 12 | 258.4 (100.0-697.3) | .029 |
| C4d (classical) | 279-1846 | 29 | 1899.9 (420.0-4717.1) | 12 | 1558.7 (546.6-4096.4) | .55 |
| C5a (terminal) | 19-48 | 29 | 45.0 (12.4-154.0) | 12 | 38.8 (24.4-48.3) | .53 |
| Markers (complement pathway) | Normal values, ng/dL | TA-TMA + GVHD | GVHD only | |||
|---|---|---|---|---|---|---|
| n | Median (range) | n | Median (range) | P | ||
| GVHD onset | ||||||
| BBPlus (alternative) | 244-961 | 21 | 403.2 (124.5-2323.0) | 14 | 481.5 (239.8-1816.6) | .87 |
| C5b-9 (terminal) | 34-248 | 21 | 289.8 (147.5-643.0) | 14 | 263.7 (169.1-518.3) | .46 |
| C4d (classical) | 279-1846 | 21 | 2854.8 (962.0-5016.2) | 14 | 2678.6 (1399.7-6205.7) | .74 |
| C5a (terminal) | 19-48 | 21 | 52.0 (13.2-105.2) | 14 | 42.5 (18.8-89.1) | .71 |
| 2-6 wk after GVHD onset | ||||||
| BBPlus (alternative) | 244-961 | 29 | 600.0 (130.6-2119.0) | 12 | 209.3 (94.5-908.7) | .0045 |
| C5b-9 (terminal) | 34-248 | 29 | 425.9 (193.2-860.0) | 12 | 258.4 (100.0-697.3) | .029 |
| C4d (classical) | 279-1846 | 29 | 1899.9 (420.0-4717.1) | 12 | 1558.7 (546.6-4096.4) | .55 |
| C5a (terminal) | 19-48 | 29 | 45.0 (12.4-154.0) | 12 | 38.8 (24.4-48.3) | .53 |
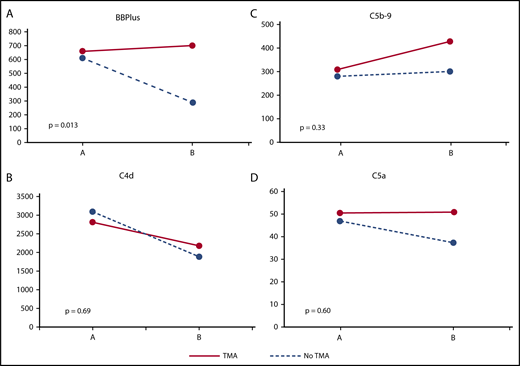
Patients with TA-TMA experience persistent activation of alternative complement pathway 2 to 6 weeks after GVHD onset. (A) BBPlus is a marker of alternative complement pathway activation. GVHD patients with and without TA-TMA had similar BBPlus levels at GVHD onset (point A on x-axis), but those who did not go on to develop TA-TMA experienced decreases in BBPlus levels over the next 2 to 6 weeks after GVHD onset (point B on x-axis). (B) C4d is a marker of classical complement pathway activation and is elevated in GVHD patients with and without TA-TMA at GVHD onset and decreases in both over time with no statistically significant difference in the change over time. No statistically significant differences were noted for changes in (C) C5b-9 or (D) C5a both markers of terminal complement pathway activation. Differential ongoing activation of the alternative complement pathway appears to be a determinant of developing TA-TMA.
Patients with TA-TMA experience persistent activation of alternative complement pathway 2 to 6 weeks after GVHD onset. (A) BBPlus is a marker of alternative complement pathway activation. GVHD patients with and without TA-TMA had similar BBPlus levels at GVHD onset (point A on x-axis), but those who did not go on to develop TA-TMA experienced decreases in BBPlus levels over the next 2 to 6 weeks after GVHD onset (point B on x-axis). (B) C4d is a marker of classical complement pathway activation and is elevated in GVHD patients with and without TA-TMA at GVHD onset and decreases in both over time with no statistically significant difference in the change over time. No statistically significant differences were noted for changes in (C) C5b-9 or (D) C5a both markers of terminal complement pathway activation. Differential ongoing activation of the alternative complement pathway appears to be a determinant of developing TA-TMA.
Patient outcomes in TA-TMA
Overall 100-day mortality rate for the study population was 13.7% (17 of 124). Of the 17 patients who died prior to post-HCT day 100, 15 had developed TA-TMA prior to death. Among patients surviving until post-HCT day 100, the 1-year OS from day 100 was 36.7% (95% CI, 21.8%-51.7%) in patients with TA-TMA prior to day 100 and 69.5% (95% CI, 57.2%-78.9%) in those without TA-TMA occurring before day 100 (P < .001; HR = 2.53; 95% CI, 1.53-4.18). OS from post-HCT day 100, dichotomized by presence or absence of TA-TMA prior to day 100, is shown in Figure 3. Overall 180-day mortality rate for the study population was 29% (36 of 124) with 91.7% of patients (33 of 36) who died having developed TA-TMA prior to death.
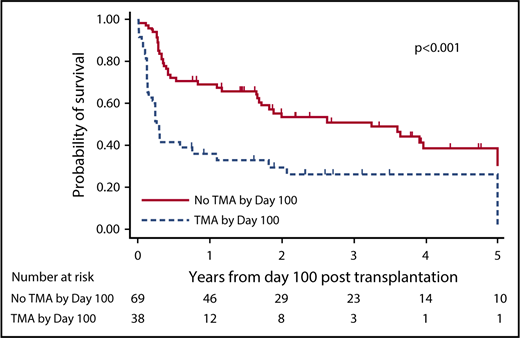
Development of TA-TMA prior to post-HCT day 100 is associated with poor OS. In patients surviving until post-HCT day 100, survival curves are shown dichotomized by patients who developed TA-TMA prior to day 100 and those who did not. There is clear separation of the curves beginning from day 100 through at least 3 years of follow-up demonstrating a significant survival advantage for patients who did not develop TA-TMA in the first 100 days after HCT.
Development of TA-TMA prior to post-HCT day 100 is associated with poor OS. In patients surviving until post-HCT day 100, survival curves are shown dichotomized by patients who developed TA-TMA prior to day 100 and those who did not. There is clear separation of the curves beginning from day 100 through at least 3 years of follow-up demonstrating a significant survival advantage for patients who did not develop TA-TMA in the first 100 days after HCT.
Patients with TA-TMA spent more time in the hospital after initial transplant admission than those with GVHD alone (median total LOS, 53 vs 25.5 days; P < .001) despite significantly shorter follow-up time in TA-TMA patients (median total follow-up, 209 vs 868 days; P < .001). Among patients with steroid-responsive GVHD, those with concomitant TA-TMA spent more time in the hospital after initial transplant admission than those with GVHD alone (median total LOS: 71 vs 22.5 days; P = .001). This was regardless of significantly shorter follow-up time in TA-TMA patients (median total follow-up, 687 vs 1147 days; P = .04). Among patients with steroid-refractory or -dependent GVHD, there was marginally significant difference in LOS based on the presence or absence of TA-TMA (median total LOS, 50 vs 27 days for TA-TMA and GVHD alone respectively; P = .07).
Discussion
In a population of patients with clinically severe GI GVHD, we have shown evidence of TA-TMA in nearly 80% of patients with steroid-refractory GVHD. We have also demonstrated that patients with steroid-refractory GVHD required more pRBC and platelet transfusions than those with steroid-dependent or -responsive disease. Regardless of concomitant GI bleeding, patients with TA-TMA required more pRBC transfusions than those without. In our time-dependent model, GVHD occurrence increased the risk of TA-TMA with the greatest risk in steroid-refractory GVHD patients. This chronology is further supported by biopsy review and complement measurement with steroid-refractory GVHD patients demonstrating some histologic evidence of TMA within a week of GVHD onset but no evidence of differential complement activation in patients with TA-TMA until 2 to 6 weeks later. The transplant-related mortality associated with steroid-refractory GVHD is roughly 50%, reflecting a need for better therapy and consideration of alternative explanations for lack of response to steroids.38,39
A link between endothelial injury and GVHD is supported by a number of recent studies.40-44 The ability to predict response to GVHD therapy using suppressor of tumorigenicity 2 (ST2) as a biomarker has been reproducibly demonstrated.22,23 In addition, a connection between endothelial injury and release of soluble ST2 has been proposed and is supported by evidence from other inflammatory states like systemic scleroderma and chronic kidney disease.45,46 The release of ST2 has been hypothesized to drive type 2 helper T (Th2) cells toward a type 1 (Th1) phenotype.23 In murine models, augmentation of the CD4+ Th1 population has led to increased severity of hepatic and intestinal GVHD.47 An association between TA-TMA and elevated ST2 has been described, suggesting an overlap between steroid-refractory GVHD and TA-TMA linked by endothelial injury, or perhaps, untreated TA-TMA contributes to the excessive mortality associated with steroid-refractory GVHD.32,35
Endothelial injury has been shown to activate complement in models of anoxic injury, purportedly via decreased expression of protectin (CD59).48,49 An increase in complement activation is to be expected in the setting of inflammation due to GVHD or other insult and we did demonstrate activation of the classical complement pathway at GVHD onset regardless of later development of TA-TMA. C4d staining of intestinal biopsies has demonstrated increased incidence of C4d deposition in the setting of acute GVHD as well as infection.50,51 Although activation of the classical pathway was similar at GVHD onset, we have also demonstrated differential activation of the alternative and terminal pathways a few weeks after GVHD onset in patients who go on to develop TA-TMA. Similar findings using a different marker of alternative pathway activation (C3b) have been recently reported.52 In combination with our histologic data, we have shown that there is local, organ-specific evidence of TMA early in the course of GI GVHD but that evidence of differential complement activation at a systemic level does not emerge until weeks later. With local GI GVHD serving as the initial complement-driving insult, patients who go on to develop TA-TMA appear to do so as a function of ongoing complement activation via the alternative complement pathway.
There are a number of clinical implications of our study. We have demonstrated the potential utility for measuring complement activation, particularly BBPlus and C5b-9, as early as 2 weeks after GVHD onset. This association should be validated in a larger, prospective study, but the identification of ongoing complement activation via the alternative pathway weeks after GVHD onset should prompt consideration for the diagnosis of concomitant TA-TMA. We have also identified a greater need for transfusion support among patients with steroid-refractory GVHD compared with less severe cases. Although confounding factors like active infection or drug side effects may contribute to cytopenias in the post-HCT setting, the significant difference in transfusion requirement demonstrated in the period immediately following GVHD onset warrants consideration of concomitant TA-TMA diagnosis in transfusion-dependent patients.
We were unable to document significant differences in TMA-associated histology findings between GVHD steroid response groups within a week of GVHD onset, though the greater prevalence of loss of glands among steroid-refractory GVHD compared with steroid-responsive GVHD patients was marginally significant. Small sample size and uncertainty about the ideal timing of biopsy to diagnose TA-TMA may have contributed to this lack of association between TMA-associated histology findings and GVHD steroid response. Despite current institutional standards recommending endoscopy at initial GI GVHD presentation, only 66 of 124 patients underwent biopsy within 7 days of symptom onset. The optimal timing for histologic assessment in suspected TA-TMA cases also remains unclear. Nonetheless, we would recommend endoscopy with biopsy at GI GVHD onset with careful attention to the proposed TMA criteria of El-Bietar et al36 with further prospective study of intestinal biopsies among patients with TA-TMA needed.
We have provided support for plasma-based testing to aid in earlier identification of TA-TMA allowing for earlier intervention. Among patients not surviving the first 100 days of HCT, the vast majority developed TA-TMA prior to death. Furthermore, patients who developed TA-TMA within the first 100 days of HCT but surviving beyond day 100 experienced excessive mortality with clear separation of survival curves through the first 3 years following HCT. Early identification and management of TA-TMA is important, not only for patient outcomes, but also in containing health care costs. Concomitant GVHD and TA-TMA was associated with longer hospital stays than GVHD alone. This difference was pronounced among patients with steroid-responsive GVHD who should, theoretically, spend less time in the hospital than steroid-refractory or -dependent patients. Furthermore, a significantly higher need for pRBC transfusions in patients with TA-TMA, regardless of concomitant GI bleed, adds significant cost in both inpatient and outpatient care settings.
We acknowledge that our study is limited by its retrospective nature. We were unable to incorporate direct antiglobulin, coagulation studies, or LDH in our diagnostic criteria due to missing data; however, we did use more stringent criteria for cytopenias and end-organ involvement than BMT-CTN or IWG criteria.1,7 In using the more strict criteria, we have likely selected for a sicker population and avoided confounding by medication side effects that can contribute to less severe post-HCT cytopenias. Despite lack of LDH inclusion in diagnostic criteria, it was found in a greater proportion of TA-TMA patients than those without. We also included subgroup analysis of patients with GI bleeding to account for the possibility of more liberal transfusion thresholds. The smaller sample size for biopsy specimen analysis may have limited our ability to detect differences in TMA-associated histology findings at GVHD onset. The lack of patient samples for measurement of plasma complement levels for the entire data set is another limitation. Small sample size may have hindered our ability to detect significant differences in complement activation between groups.
In addition to diagnosis of GVHD, fungal and viral infections were associated with a greater risk of developing TA-TMA, potentially via endothelial injury and subsequent complement activation. As our study was limited to patients with GVHD, we cannot evaluate the roles of infection or other non-GVHD insults and their association with TA-TMA in transplant recipients who do not develop GVHD. Future studies seeking to disentangle the complex relationships between TA-TMA, GVHD, infection, and toxicities of the drugs used to treat and prevent these conditions are needed. Patients with GVHD, especially steroid-refractory disease, are at high risk of infection.53 On the other hand, bacterial infection in particular may precede GVHD onset and has been shown to increase the risk of developing clinically significant GVHD.54,55 Relevant to both prophylaxis and treatment of GVHD, many retrospective and prospective studies have identified the use of sirolimus and calcineurin inhibitors as risk factors for developing TA-TMA.1,4,56 The effect of cytokine inhibition and other second-line GVHD therapies on risk of TA-TMA is unknown and warrants further study.
In the future, we will seek to further explore the overlap between GVHD and TA-TMA. Prognostic biomarkers of GVHD, especially ST2 as a marker of steroid-refractoriness, warrant further study in patients with TA-TMA. We suspect that GVHD-associated endothelial injury leads to TA-TMA via unregulated complement activation. The inability to curb complement activation may be due to deletions in genes encoding complement regulatory factors. The incidence of genetic susceptibilities to complement-associated TMAs has been described in aHUS, pediatric TA-TMA, and the general population but warrants further study in the adult HCT population.57
In conclusion, we have demonstrated a strong relationship between steroid-refractory GVHD and TA-TMA. We have also shown that GVHD almost always precedes the diagnosis of TA-TMA. We have proposed a mechanistic link between GVHD, endothelial injury, complement activation, and TA-TMA based on our demonstration of the temporal relationship between GVHD and TA-TMA. Early organ-specific evidence of TA-TMA from GI biopsies, particularly in steroid-refractory GVHD, led to later systemic evidence of increased alternative and terminal complement pathway activation in patients who go on to develop TA-TMA. It is important to consider a diagnosis of TA-TMA in patients with GI GVHD who require transfusion support and to communicate concern for TA-TMA to consulting gastroenterologists and pathologists as histologic evidence of TMA may co-occur with GVHD onset and precede systemic signs of TA-TMA. Larger prospective studies are necessary to explore differential activation of complement pathways, particularly the alternative pathway, in both TA-TMA and GVHD and whether complement blockade is an effective therapeutic strategy in patients with both GVHD and TA-TMA. Future studies of ST2 or other markers of endothelial injury may provide evidence of the proposed mechanistic link between GVHD, endothelial injury, and TA-TMA.
The full-text version of this article contains a data supplement.
Acknowledgment
This work was supported in part by funding from National Institutes of Health, National Cancer Institute grant 4T32CA165998-05 (S.A.W. and S.D.).
Authorship
Contribution: S.A.W. and S.V. conceived of the study; Q.Z. and A.A. performed statistical analysis; S.A.W., S.V., Q.Z., and S.C. interpreted the data; M.Y. and L.B. reviewed biopsy specimens; P.R. provided the plasma samples for correlative study; S.Y. and H.W. performed complement marker measurement; S.A.W. and M.B. collected patient data; and S.A.W., Q.Z., S.J., J.E.B., B.W., H.C., A.S.M., Y.E., S.P., S.D., S.M.D., and S.V. wrote and edited the manuscript.
Conflict-of-interest disclosure: S.C. has received research funding and honoraria from Alexion, Inc, the manufacturer of eculizumab. The remaining authors declare no competing financial interests.
Correspondence: Sumithira Vasu, Division of Hematology, Department of Internal Medicine, The Ohio State University, B310 Starling-Loving Hall, 320 W 10th Ave, Columbus, OH 43210; e-mail: sumithira.vasu@osumc.edu.

