

Key Points
Immunoproteasome inhibitor ONX-0914 has antimyeloma activity in vitro similar to that of bortezomib and carfilzomib.
LU-102 sensitizes MM cells to a pharmacologically relevant concentration of ONX-0914, which synergizes with bortezomib in vivo.

Abstract
Proteasome inhibitors bortezomib, carfilzomib and ixazomib (approved by the US Food and Drug Administration [FDA]) induce remissions in patients with multiple myeloma (MM), but most patients eventually become resistant. MM and other hematologic malignancies express ubiquitous constitutive proteasomes and lymphoid tissue–specific immunoproteasomes; immunoproteasome expression is increased in resistant patients. Immunoproteasomes contain 3 distinct pairs of active sites, β5i, β1i, and β2i, which are different from their constitutive β5c, β1c, and β2c counterparts. Bortezomib and carfilzomib block β5c and β5i sites. We report here that pharmacologically relevant concentrations of β5i-specific inhibitor ONX-0914 show cytotoxicity in MM cell lines similar to that of carfilzomib and bortezomib. In addition, increasing immunoproteasome expression by interferon-γ increases sensitivity to ONX-0914 but not to carfilzomib. LU-102, an inhibitor of β2 sites, dramatically sensitizes MM cell lines and primary cells to ONX-0914. ONX-0914 synergizes with all FDA-approved proteasome inhibitors in MM in vitro and in vivo. Thus, immunoproteasome inhibitors, currently in clinical trials for the treatment of autoimmune diseases, should also be considered for the treatment of MM.
Introduction
Multiple myeloma (MM) is a plasma cell malignancy with a median survival of approximately 7 years.1,2 The proteasome inhibitors bortezomib, carfilzomib, and ixazomib are critical components of MM treatment regimens. Although initial response rates with these treatments are high, the majority of patients eventually become resistant and succumb to the disease, underlining a need for new treatments.
MM cells express constitutive proteasomes and immunoproteasomes. The constitutive proteasomes are expressed ubiquitously through the body, whereas immunoproteasomes are highly expressed in cells and tissues of the immune system3,4 and constitute 30% to 90% of the total proteasomes in MM cells.5,6 A decrease in the expression of immunoproteasomes correlates with a better response to bortezomib in patients,7 whereas patients with relapsed/refractory MM show increased immunoproteasome activity.8 Thus, immunoproteasome inhibition may be the key to increasing proteasome inhibition (and thereby enhancing MM cell death) in patients with relapsed/refractory MM.
Both types of proteasomes have 3 pairs of active sites, the β5c (PSMB5, or chymotrypsin-like), β1c (PSMB6, or caspase-like), and the β2c (PSMB7, or trypsin-like) sites in the constitutive proteasome and the β5i (PSMB8, LMP7), β1i (PSMB9, LMP2), and the β2i (PSMB10, LMP10, MECL1) sites in the immunoproteasome.9 Bortezomib, carfilzomib and ixazomib are potent inhibitors of the β5i and β5c sites. Previous studies have shown that specific inhibition of β5i is not sufficient to kill myeloma cells6 ; however, it is not known whether coinhibition of β1i and β2i sites will make an inhibitor cytotoxic. We developed LU-102, a specific cell-permeable inhibitor of β2 sites and found that it dramatically sensitizes myeloma and solid tumor cells to bortezomib and carfilzomib,10,11 restores sensitivity of primary cells from bortezomib- and carfilzomib-resistant myeloma patients to these agents, and synergizes with carfilzomib in murine models of myeloma.12 Combined inhibition of β5 and β2 sites may increase mechanism-based toxicity (eg, gastrointestinal, renal, and cardiac toxicities) because of increased inhibition of protein breakdown in these tissues; however, replacing bortezomib and carfilzomib with an immunoproteasome inhibitor should reduce these adverse effects because expression of immunoproteasomes in these tissues is low. Mice lacking all 3 immunoproteasomes subunits are viable.13
Although several immunoproteasome inhibitors have been tested against MM cells, their specificity at active doses was not confirmed,5,14-17 and the effect of combined inhibition of β5i and β2 sites has not been explored. β5i inhibitor ONX-0914 (PR-957) has in vivo activity in murine models of many different autoimmune disorders,18-22 in which selective killing of auto-antibody plasma cells is believed to contribute to efficacy. A derivative of ONX-0914, KZR-616, is currently being developed for the treatment of autoimmune diseases. It recently completed a successful phase 1a study in healthy volunteers (ACTRN12616001040459)23 and seems to bypass the toxic effects (eg, gastrointestinal toxicities, neurotoxicity). Therefore, the possibility of repositioning KZR-616 for the treatment of MM must be explored.
KZR-616 is not yet available to us; therefore, we used ONX-0914 as a compound tool to determine of feasibility of inhibition immunoproteasomes for the treatment of MM. Here, we demonstrate that MM cells, which are sensitive to clinically relevant concentrations of carfilzomib and bortezomib, also respond to treatment with pharmacologically relevant concentrations of ONX-0914. Furthermore, we demonstrate that subtoxic doses of the β2 inhibitor LU-102 strongly sensitize MM cells to ONX-0914, as observed previously for bortezomib and carfilzomib.11,12,24 We also found that ONX-0914 synergizes with inhibitors approved by the US Food and Drug Administration (FDA) in vitro as well as in vivo.
Materials and methods
Cell lines
KMS-11 cells were purchased from the Health Science Research Resources Bank (Osaka, Japan). NCI-H929 and RPMI-8226 cells were purchased from the American Type Culture Collection. All other cells lines were authenticated by the University of Vermont Human Cell Line Authentication Facility. Cells were cultured in RPMI-1640 (Hyclone) medium supplemented with 10% fetal bovine serum (Hyclone), penicillin (100 μg/mL), streptomycin (100 U/mL), and the antimycoplasma antibiotic Plasmocin (1.5 μg/mL; InvivoGen, San Diego, CA). Patient samples were collected in accordance with the Declaration of Helsinki and under the protocol approved by the Dartmouth College Committee on Protection of Human Subjects or approved by the Dana-Farber Cancer Institute Committee on Protection of Human Subjects, and all animal experiments were conducted according to the protocol approved by Dartmouth Institutional Animal Care and Use Committee.
Cell-based assays
Bortezomib, carfilzomib, and ixazomib were purchased from LC Laboratories, and ONX-0914 was purchased from MedKoo. LU-102 was synthesized as described.11 Unless otherwise stated, cell lines were treated with carfilzomib, bortezomib, ONX-0914, and ixazomib for 1 hour and then cultured in the presence or absence of LU-102 for 47 hours; viability was then assessed with Alamar Blue mitochondrial dye conversion assay (Invitrogen or Bio-Rad) as described.25 For flow cytometry, CD138-PE (BD Pharmingen #550805) and Zombie Aqua Fixable Viability Kit (BioLegend #423101) were used. The values on all graphs are averages ± standard error of the mean of a number of experiments (supplemental Table 1). At least 2 biological replicates were averaged to define the 4-parameter fit of concentration-dependence curves used to determine the half maximal inhibitory concentration.
Cells were lysed with 0.05% digitonin in 50 mM tris(hydroxymethyl)aminomethane (pH 7.5), 250 mM sucrose, 5 mM MgCl2, 1 mM adenosine triphosphate, 0.5 mM EDTA, and 1 mM dithiothreitol. To assay activity of β5i, β5c, β1i, and β1c subunits, 10 µg of protein were coincubated with 2.5 µM BODIPY-NC-001 and BODIPY-NC-005-VS (compounds 23 and 24 in Verdoes et al26 ) for 1 hour at 37°C. To resolve β2, β2i, β1, and β1i subunits, extracts were co-treated with MV-15127 and BODIPY-NC-001.26 Proteasome subunits were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 0.75-mm-thick 12.5% gels and a Laemmli buffer system. Probe-modified subunits were revealed by fluorescent imaging. Gels were then stained with Coomassie blue dye as a loading control. β5i and β5c activity was determined using the 7-amino-4-methylcoumarin amide (amc) substrates Suc-LLVY-amc (β5c + β5i, 100 μM) and Ac-ANW-amc (β5i, 200 μM)28 as described.24,29
In vivo testing
Male NSG (Nod/Scid/IL-2 receptor γ-chain knockout; The Jackson Laboratory) mice were treated with 100 cgy of radiation (from a cesium irradiator; sublethal dose) based on Mitsiades et al.30 The following day, 2 million MM1.S cells in phosphate-buffered saline were injected into these mice via the tail vein. Bortezomib (0.5 mg/kg) and ONX-0914 (up to 15 mg/kg) were dissolved in 10 mM calcium citrate (pH 6), with 10% Captisol and injected subcutaneously 2 times per week (as indicated in Figure 4D). Animals were euthanized when they began dragging a hind leg. Blood samples were taken, and human immunoglobulin G λ levels were determined by western blot using anti-human λ-chain antibody (Vector, #AI-33070, 1:500), with a fluorescently labeled anti-goat secondary antibody (Rockland, #605-745-002, 1:10000), and they were imaged on an Odyssey (LICOR) scanner.
Results
ONX-0914 is cytotoxic to MM cells that express high levels of β5i
To determine whether the immunoproteasome content in cultured MM cells is similar to that of primary MM cells in which 30% to 90% of proteasomes are immunoproteasomes,5 we treated extracts with activity-based probes (ie, fluorescently labeled irreversible inhibitors) BODIPY-NC-005-vs and BODIPY-NC-001,26 and then resolved the fluorescently labeled subunits on an sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel (Figure 1A). Most MM cells have almost equal amounts of active β5i and β5c (MM1.S, U266, RPMI-8226) or have a greater amount of β5c (H929, KMS-11, KMS-18, KMS-21-PE, LR5). Thus, cultured MM cells express immunoproteasomes, although they have slightly lower β5i:β5c ratios than the primary myeloma cells reported in the literature5 or that we isolated from patients (Figure 1B).
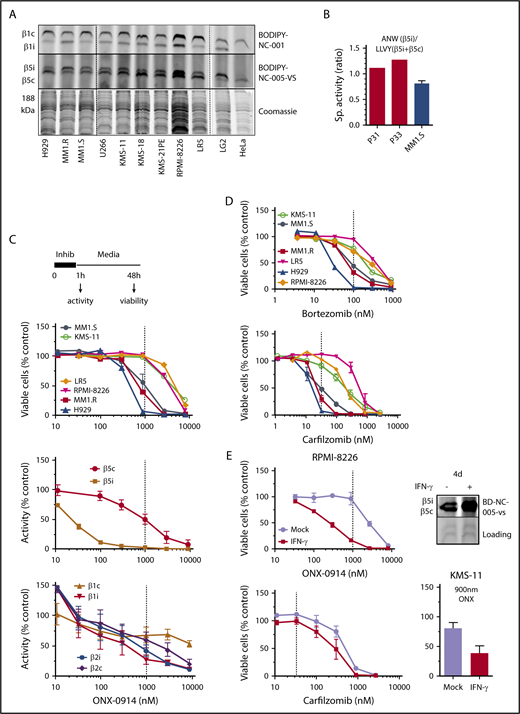
Effect of ONX-0914 on MM cells. (A) Expression of immunoproteasomes in MM cell lines as determined by the activity-based probes. Immortalized B-cell line LG2, which expresses high levels of immunoproteasomes, and HeLa cells, which express little or no immunoproteasomes were used as references (n = 2). (B) Cleavage rates of β5i-specific substrate Ac-ANW-amc and of β5c and β5i substrate Suc-LLVY-amc were measured in extracts of primary MM cells isolated from relapsed/refractory patients using EasySep Human CD138 Positive Selection Kit II (STEMCELL Technologies, Catalog #18357), and ANW:LLVY ratio was used as readout of relative β5i expression. (C) ONX-0914 is cytotoxic to MM cells. Top: cells were treated with ONX-0914 for 1 hour followed by Alamar Blue assay 47 hours later (n = 2-3; see supplemental Table 1 for exact number of cells for each cell line). In a parallel experiment, proteasome inhibition was measured by site-specific fluorogenic substrates (middle graph) or activity-based probes (bottom graph) immediately after 1-hour treatment with ONX-0914 (n = 4). Dashed line indicates in vivo relevant inhibition (eg, concentration that causes ≥95% of β5i and ∼50% of β5c inhibition similar to maximal tolerated dose in vivo18 ). (D) Clinically relevant concentrations of bortezomib or carfilzomib (n = 2-3) have cytotoxicity similar to that of ONX-0914. Dashed lines indicate clinically relevant doses. Except for KMS-11 (n = 5), bortezomib data are from Figure 1 of Shabaneh et al.25 (E) IFN-γ treatment increases the sensitivity of MM cells to ONX-0914, but not to carfilzomib. Cells were treated with IFN-γ for 5 days and then treated with ONX-0914 (upper left and bottom right) or carfilzomib (bottom left) as in panel C. Upper right panel shows increase in β5i activity after 4 days as measured by BODIPY-NC-005-VS (n = 2-3). Sp., specific.
Effect of ONX-0914 on MM cells. (A) Expression of immunoproteasomes in MM cell lines as determined by the activity-based probes. Immortalized B-cell line LG2, which expresses high levels of immunoproteasomes, and HeLa cells, which express little or no immunoproteasomes were used as references (n = 2). (B) Cleavage rates of β5i-specific substrate Ac-ANW-amc and of β5c and β5i substrate Suc-LLVY-amc were measured in extracts of primary MM cells isolated from relapsed/refractory patients using EasySep Human CD138 Positive Selection Kit II (STEMCELL Technologies, Catalog #18357), and ANW:LLVY ratio was used as readout of relative β5i expression. (C) ONX-0914 is cytotoxic to MM cells. Top: cells were treated with ONX-0914 for 1 hour followed by Alamar Blue assay 47 hours later (n = 2-3; see supplemental Table 1 for exact number of cells for each cell line). In a parallel experiment, proteasome inhibition was measured by site-specific fluorogenic substrates (middle graph) or activity-based probes (bottom graph) immediately after 1-hour treatment with ONX-0914 (n = 4). Dashed line indicates in vivo relevant inhibition (eg, concentration that causes ≥95% of β5i and ∼50% of β5c inhibition similar to maximal tolerated dose in vivo18 ). (D) Clinically relevant concentrations of bortezomib or carfilzomib (n = 2-3) have cytotoxicity similar to that of ONX-0914. Dashed lines indicate clinically relevant doses. Except for KMS-11 (n = 5), bortezomib data are from Figure 1 of Shabaneh et al.25 (E) IFN-γ treatment increases the sensitivity of MM cells to ONX-0914, but not to carfilzomib. Cells were treated with IFN-γ for 5 days and then treated with ONX-0914 (upper left and bottom right) or carfilzomib (bottom left) as in panel C. Upper right panel shows increase in β5i activity after 4 days as measured by BODIPY-NC-005-VS (n = 2-3). Sp., specific.
We then pulse-treated a panel of 6 MM cells with ONX-0914 for 1 hour and determined their viability 48 hours later (Figure 1C). The pulse treatment better mimics the bolus injections currently used in patients than the continuous exposure used in most in vitro studies.25 In parallel experiments, we assessed the extent to which ONX-0914 inhibits the different active sites in MM1.S cells (Figure 1C). Although β5i specific at low concentrations (eg, 100 nM), this agent inhibited other subunits of immune and constitutive proteasomes at higher concentrations. ONX-0914 (1 μM) inhibited proteasomes to the same degree as it did in the tissues of treated mice at the maximum tolerated dose.18 This represents complete inhibition of β5i and ∼50% inhibition of β5c, β1i, and β1c (Figure 1C). Therefore, for the remainder of the study, we considered 1 μM ONX-0914 to be a pharmacologically relevant concentration. This concentration was cytotoxic to several cell lines. We have previously reported that a 1-hour pulse with 100 nM bortezomib leads to a clinically achievable 75% inhibition of β5 subunits25 and that 33 nM carfilzomib causes the clinically achievable ∼90% β5 inhibition.24 As with ONX-0914, bortezomib and carfilzomib reduced the viability of MM1.S, MM1.R, and H929 cells, but not of any other cell lines at these clinically relevant concentrations (Figure 1D). Thus, at relevant doses, ONX-0914 has in vitro cytotoxicity similar to that of carfilzomib and bortezomib.
Because primary cells have a higher β5i:β5c ratio (Figure 1B), we tested whether increasing the expression of β5i by interferon-γ (IFN-γ) specifically enhances sensitivity to an immunoproteasome inhibitor. Indeed, IFN-γ treatment increased cytotoxicity to ONX-0914, but not to carfilzomib (Figure 1E), indicating that increased expression of immunoproteasomes enhances sensitivity to immunoproteasome inhibitors.
Cells upregulate β5c as they recover from β5i inhibition
With the exception of the few most sensitive MM cell lines, proteasome activity in MM cells recovers after treatment with clinically relevant concentrations of bortezomib.25 A 1-hour pulse treatment of RPMI-8226 cells with subtoxic levels (1 µM) of ONX-0914 or with 100 nM carfilzomib (Figure 2A) causes a similar recovery of total β5 activity (eg, combined β5i and β5c activity) within 18 hours (Figure 2B-C). This recovery is mediated by the transcription factor Nrf1,31,32 which regulates the transcription of all constitutive proteasome subunits, but not of β5i (PSMB8) and other immunoproteasome subunits.33 We conclude that β5c-dependent recovery of proteasome activity can desensitize MM cells to β5i inhibitors, suggesting that an agent that blocks recovery should be able to increase the efficacy of immunoproteasome inhibitors.
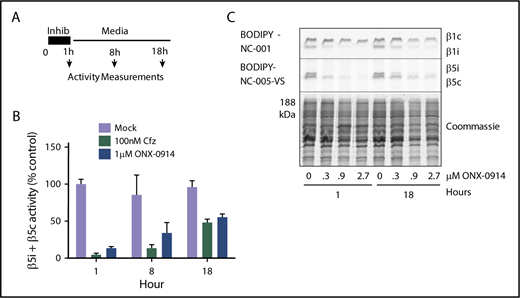
ONX-0914 treated cells upregulate β5c. (A) RPMI-8226 cells were treated as shown. At times indicated, either total β5 (β5i + β5c) activity was measured with Suc-LLVY-amc (n = 2) (B) or individual subunits were measured with ABPs (n = 2) (C).
ONX-0914 treated cells upregulate β5c. (A) RPMI-8226 cells were treated as shown. At times indicated, either total β5 (β5i + β5c) activity was measured with Suc-LLVY-amc (n = 2) (B) or individual subunits were measured with ABPs (n = 2) (C).
LU-102 sensitizes cells to clinically relevant concentrations of ONX-0914
We have shown that LU-102, the β2c and β2i inhibitor, dramatically sensitizes MM cells to bortezomib and carfilzomib,11,12 and that the mechanism of such sensitization involves inhibition of the Nrf1-dependent recovery of proteasome activity.10 On the basis of this phenomenon, we next tested whether LU-102 sensitizes cells to ONX-0914. When used as a single agent, LU-102 did not cause cell death at β2c/β2i-specific concentrations (Figure 3A-B), except in the H929 cells that are highly sensitive to all proteasome inhibitors. However, subtoxic β2-specifc concentrations of LU-102 dramatically sensitized MM cell lines to ONX-0914 (Figure 3C-D; supplemental Figure 1A), decreasing the half maximal inhibitory concentration by threefold to eightfold (Figure 3E) and reducing viability of all cell lines treated for 1 hour with the pharmacologically relevant 0.9 µM ONX-0914 (Figure 3F). LU-102–induced decrease in viability was similar in magnitude to the reduction seen in carfilzomib-treated cells (supplemental Figure 1). The cells treated with IFN-γ were especially sensitive to the combined treatments. Importantly, LU-102 synergized with ONX-0914 in inducing death of CD138+ cells from MM patients (Figure 3G). As expected, LU-102 blocked the recovery of β5 activity after treatment with either carfilzomib or ONX-0914 (Figure 3H). Thus, LU-102 is a potent sensitizer of MM cells to ONX-0914.
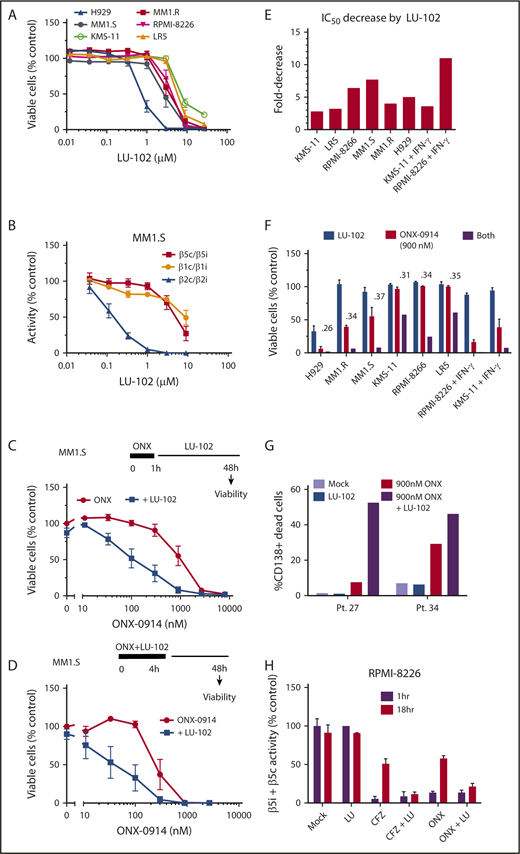
Addition of LU-102 sensitizes cells to in vivo relevant concentrations of ONX-0914. (A) Effect of 48-hour treatment with LU-102 on viability MM cells as determined by Alamar Blue (n = 2-4). (B) Inhibition of proteasome in MM1.S cells after 2-hour treatment as determined by the Proteasome-Glo assay (n = 3) (Promega). (C) MM1.S cells were treated with ONX-0914 for 1 hour, and then treated with 1 μM LU-102 for 47 hours (n = 3). (D) MM1.S cells were cotreated for 4 hours with ONX-0914 and 3 μM LU-102 (n = 2). (E) Cells were treated by ONX-0914, followed by LU-102 (250 nM in H929 and MM1.R, 1 μM in all others) for 47 hours (n = 2-6), and half maximal inhibitory concentrations of MM cells was determined form dose-response curves (supplemental Figure 1). (F) Viability of cells treated with 900 nM ONX-0914 in experiment shown in panel E. Numbers are combination indexes (CIs). (G) Primary cells isolated from plural effusions of bortezomib-refractory patients were treated with 0.9 µM ONX-0914 for 4 hours with or without 2 μM LU-102, and viability of CD138+ cells was determined by flow cytometry at 48 hours using PE Mouse Anti-Human CD138 and Zombie Aqua Fixable Viability antibodies. (H) LU-102 blocks the recovery of β5i/β5c activity over time after 1-hour treatment. Total β5 activity was measured with Proteasome-Glo in RPMI-8226 cells, either immediately after 1-hour treatment with 1 µM ONX-0914 or 100 nM carfilzomib or after a 17-hour recovery in the presence or absence of 1 µM LU-102 (n = 2). Pt., patient.
Addition of LU-102 sensitizes cells to in vivo relevant concentrations of ONX-0914. (A) Effect of 48-hour treatment with LU-102 on viability MM cells as determined by Alamar Blue (n = 2-4). (B) Inhibition of proteasome in MM1.S cells after 2-hour treatment as determined by the Proteasome-Glo assay (n = 3) (Promega). (C) MM1.S cells were treated with ONX-0914 for 1 hour, and then treated with 1 μM LU-102 for 47 hours (n = 3). (D) MM1.S cells were cotreated for 4 hours with ONX-0914 and 3 μM LU-102 (n = 2). (E) Cells were treated by ONX-0914, followed by LU-102 (250 nM in H929 and MM1.R, 1 μM in all others) for 47 hours (n = 2-6), and half maximal inhibitory concentrations of MM cells was determined form dose-response curves (supplemental Figure 1). (F) Viability of cells treated with 900 nM ONX-0914 in experiment shown in panel E. Numbers are combination indexes (CIs). (G) Primary cells isolated from plural effusions of bortezomib-refractory patients were treated with 0.9 µM ONX-0914 for 4 hours with or without 2 μM LU-102, and viability of CD138+ cells was determined by flow cytometry at 48 hours using PE Mouse Anti-Human CD138 and Zombie Aqua Fixable Viability antibodies. (H) LU-102 blocks the recovery of β5i/β5c activity over time after 1-hour treatment. Total β5 activity was measured with Proteasome-Glo in RPMI-8226 cells, either immediately after 1-hour treatment with 1 µM ONX-0914 or 100 nM carfilzomib or after a 17-hour recovery in the presence or absence of 1 µM LU-102 (n = 2). Pt., patient.
Addition of ONX-0914 to FDA-approved inhibitors overcomes resistance
Our previous data indicate that inhibition of the β5i site is most effective when β5c sites are also partially inhibited. These conditions are achieved by combining ONX-0914 with lower subtoxic doses of carfilzomib or bortezomib. Because such combinations can be introduced into clinical trials more rapidly than LU-102, we combined ONX-0914 and the FDA-approved proteasome inhibitors. ONX-0914 synergized with bortezomib, carfilzomib, and ixazomib in MM1.S (Figure 4A) and RPMI-8226 (Figure 4B) cell lines.
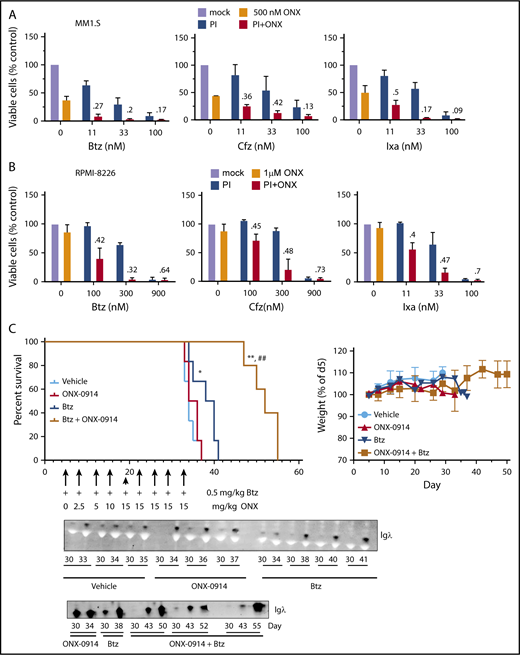
ONX-0914 is synergistic with FDA-approved inhibitors. (A-B) ONX-0914 is synergistic with bortezomib (Btz), carfilzomib (Cfz), and ixazomib (Ixa) in MM1.S (A) and RPMI-8226 (B) cells (n = 2). Numbers on the graph are CIs. (C) ONX-0914 and bortezomib have synergistic activity in orthotopic mouse model of MM. NSG mice (6 per group) were injected intravenously with MM1.S cells and then treated as shown. Weight at euthanasia is not included on the right graph because animals lose weight as a result of disease progression. Error bars indicate standard error of the mean. Bottom: 1 µL of serum samples was assayed for human Igλ by western blot. The latest sample was taken at euthanasia. *P < .05 when compared with vehicle group; **P < .005 when compared with vehicle group; ##P < .005 when compared with bortezomib only group. PI, proteasome inhibitor.
ONX-0914 is synergistic with FDA-approved inhibitors. (A-B) ONX-0914 is synergistic with bortezomib (Btz), carfilzomib (Cfz), and ixazomib (Ixa) in MM1.S (A) and RPMI-8226 (B) cells (n = 2). Numbers on the graph are CIs. (C) ONX-0914 and bortezomib have synergistic activity in orthotopic mouse model of MM. NSG mice (6 per group) were injected intravenously with MM1.S cells and then treated as shown. Weight at euthanasia is not included on the right graph because animals lose weight as a result of disease progression. Error bars indicate standard error of the mean. Bottom: 1 µL of serum samples was assayed for human Igλ by western blot. The latest sample was taken at euthanasia. *P < .05 when compared with vehicle group; **P < .005 when compared with vehicle group; ##P < .005 when compared with bortezomib only group. PI, proteasome inhibitor.
We decided to determine whether this synergistic interaction can be recapitulated in an orthotopic murine model of MM, in which MM cells form tumors in bone marrow if they are injected via the tail vein into irradiated immunodeficient mice.30 Bortezomib was chosen over carfilzomib for this study because initial clinical trials are performed in heavily pretreated patients, and phase 1 trial candidates are more likely to be refractory to bortezomib than to carfilzomib. The onset of paraplegia and the presence of immunoglobulin G λ-light chain levels in the serum were used as readouts for tumor growth. Bortezomib treatment alone slightly but significantly increased the overall survival in the experimental model compared with vehicle-injected mice (Figure 4C). Although ONX-0914 treatment alone did not increase survival, the combination of ONX-0914 and bortezomib delayed the appearance of λ-light chains in blood and significantly increased the overall survival compared with the vehicle group and the bortezomib-only group without obvious signs of additional toxicity. Thus, ONX-0914 synergizes with bortezomib in vivo.
Discussion
Our data provide a strong rationale for the use of a combination therapy that includes immunoproteasomes and β2 inhibitors. ONX-0914 is the precursor of KZR-616, which is currently being developed for the treatment of autoimmune disorders and for which a phase 1 trial in healthy volunteers has been successfully completed.23 Although KZR-616 was not available to us, the active site inhibition profile for it resembles that for ONX-0914, which has been used in our work (Christopher Kirk, Kezar Life Sciences, oral communication, 18 April 2017). Because β2 inhibitors are at the early stages of preclinical development, an increase in overall survival of mice treated with bortezomib and ONX-0914 suggests that combinations of KZ-616 with one of the FDA-approved proteasome inhibitors will yield the fastest route to the clinic.
To successfully translate these findings in the clinic, upregulation of constitutive proteasomes in response to β5i inhibition, which we detected independently (Figure 2) and which 2 other laboratories also detected,17,34 must be addressed. We suggest that immunoproteasome inhibitors should be combined with agents that, like LU-102, block the recovery of activity. Expression of constitutive proteasomes is upregulated by Nrf1. Nrf1 activation involves ubiquitin, VCP ATPase, and proteolytic activation by DDI2, an aspartic protease similar to HIV protease.31,32,35-37 Inhibitors of VCP138 and of E1 ubiquitin-activating enzyme39 are undergoing clinical trials, and it should not be difficult to develop DDI2 inhibitors.
The content of immunoproteasomes varies among patients with MM,5 and we clearly show that increased immunoproteasome expression selectively sensitizes cells to immunoproteasome inhibitors. Therefore, all clinical trials of immunoproteasome inhibitors in myeloma or hematologic malignancies should involve patient stratification. In addition to the enzyme-linked immunosorbent assay–based ProCISE assay developed by ONYX,6 we have developed several subunit-specific assays26,40 that can be adopted for clinical use.
In summary, this work provides a strong rationale for the development of immunoproteasome inhibitors in the treatment of patients with MM and also raises the possibility that these agents will be of therapeutic value for the treatment of other hematologic malignancies that also express high levels of immunoproteasomes.41,42
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Mouse Modeling Shared Resource of the Norris Cotton Cancer Center for technical assistance with animal experiments and Lenka Besse for the critical reading of the manuscript. H.S.O. and B.I.F. acknowledge ChemAxon for kindly providing the Instant JChem software to manage our compound library.
This work was supported by the Norris Cotton Cancer Center at Dartmouth College with a Prouty Pilot grant and with National Cancer Institute, National Institutes of Health (NIH) Cancer Center Support Grant 5P30 CA023108-38 (A.F.K.). S.D.-K. was supported by Rosaline Borison Memorial Fund Predoctoral Fellowship at Dartmouth. The work performed by S.D.-K. in the Mitsiades Laboratory and was supported by NIH, National Cancer Institute grants R01 CA050947 and CA196664 (C.S.M.); the de Gunzburg Myeloma Research Fund (C.S.M.); the Leukemia and Lymphoma Society (LLS) Translational Research Program and LLS Scholar Award (C.S.M.); and the Multiple Myeloma Research Foundation.
Authorship
Contribution: S.D.-K., B.I.F., C.D., H.S.O., and A.F.K. designed the research; S.D.-K., E.W.D., and A.F.K. performed the research; S.D.-K., E.W.D., B.I.F., C.D., H.S.O., and A.F.K. analyzed the data; S.D.-K., B.I.F., and A.F.K. wrote the paper; and A.B., M.G., P.G.R., and C.S.M. provided patient samples.
Conflict-of-interest disclosure: A.F.K is a Founder and Chief Scientific Officer of InhiProt. C.S.M. received research funding from Novartis, Janssen/Johnson & Johnson, TEVA, Ono, and AbbVie and has a family member employed by Takeda. P.G.R. has served on the advisory board of or received research funding from Millennium/Takeda, Celgene, Novartis, Johnson & Johnson, Oncopeptides AB, and Bristol-Myers Squibb. The remaining authors declare no competing financial interests.
Correspondence: Alexei F. Kisselev, Auburn University, Pharmacy Research Building, 720 S Donahue Dr, Auburn, AL 36849; e-mail: afk0006@auburn.edu.

