

Key Points
Mll-Af4 and mutant N-Ras cooperate to generate an aggressive B-acute lymphoblastic leukemia murine model, recapitulating human disease.
Combination MEK and ATR inhibitor treatment of Mll-Af4/N-Ras leukemia induces synergistic antileukemic effects in vitro and in vivo.

Abstract
Infant B-cell acute lymphoblastic leukemias (B-ALLs) that harbor MLL-AF4 rearrangements are associated with a poor prognosis. One important obstacle to progress for this patient population is the lack of immunocompetent models that faithfully recapitulate the short latency and aggressiveness of this disease. Recent whole-genome sequencing of MLL-AF4 B-ALL samples revealed a high frequency of activating RAS mutations; however, single-agent targeting of downstream effectors of the RAS pathway in these mutated MLL-r B-ALLs has demonstrated limited and nondurable antileukemic effects. Here, we demonstrate that the expression of activating mutant N-RasG12D cooperates with Mll-Af4 to generate a highly aggressive serially transplantable B-ALL in mice. We used our novel mouse model to test the sensitivity of Mll-Af4/N-RasG12D leukemia to small molecule inhibitors and found potent and synergistic preclinical efficacy of dual targeting of the Mek and Atr pathways in mouse- and patient-derived xenografts with both mutations in vivo, suggesting this combination as an attractive therapeutic opportunity that might be used to treat patients with these mutations. Our studies indicate that this mouse model of Mll-Af4/N-Ras B-ALL is a powerful tool to explore the molecular and genetic pathogenesis of this disease subtype, as well as a preclinical discovery platform for novel therapeutic strategies.
Introduction
More than 70% of cases of infant (<1-year old) B-cell acute lymphoblastic leukemia (B-ALL) are characterized by the translocation of the MLL1 locus with several fusion partners, most prominent among them, AF4, which accounts for ∼50% of cases.1 These leukemias are considered to have poor prognosis and have a 5-year disease-free survival ∼60%.1 However, models that faithfully recapitulate the highly aggressive nature of MLL-AF4 B-ALLs remain difficult to generate. Genetically engineered mouse models (GEMMs) that express Mll-Af4 only develop disease after long latencies2,3 or induce acute myeloid and lymphoblastic leukemias.4 More recently, retroviral transduction of cord blood–derived human CD34+ hematopoietic stem cells with human MLL fused to murine Af4 was shown to be sufficient for generating a model of pro–B-ALL in immunodeficient mice. However, when murine hematopoietic stem and progenitor cells were transduced with the same high-titer retrovirus, they generated acute myeloid leukemia in congenic mice.5 Thus, GEMMs that reflect the clinical and pathological features of MLL-rearranged (MLL-r) B-ALL and maintain species and lineage fidelity in immunocompetent backgrounds remain elusive.
Multiple sequencing studies have indicated that MLL-r B-ALLs contain a relatively low frequency of additional somatic mutations.6-10 However, there are recurrent mutations found in MLL-r acute lymphoblastic leukemia (ALL) that likely influence disease progression.7-9 Large-scale whole-genome sequencing of infant and pediatric leukemias bearing MLL rearrangements suggests that the combination of mutant RAS and MLL-AF4 is of particular interest, because they seem to co-occur at a frequency close to 50% (either activating mutations in N-RAS or K-RAS).6-10 Although one study has suggested that mutant Ras and Mll-Af4 can cooperate to accelerate disease,11 murine models that faithfully and exclusively generate B-ALLs with short latencies are still lacking.
Among MLL-r infant leukemias, those harboring mutant RAS mutations have even worse overall prognosis12 and increased resistance to standard glucocorticoid therapies (eg, prednisolone, dexamethasone).10,13,14 These observations suggest that activating RAS mutations contribute to the pathogenesis of this disease and could be an attractive therapeutic target. Several studies have demonstrated some single-agent efficacy of MEK inhibitors on MLL-r B-ALL cell lines in vitro and primary patient samples harboring activating RAS mutations ex vivo in inducing leukemia cytotoxicity.13-15 Furthermore, these studies demonstrate that the use of MEK inhibitors can partially restore sensitivity of glucocorticoid-resistant MLL-r B-ALLs to prednisolone.13,14 Although MEK inhibition did decrease cell viability, these responses were nondurable and not curative. These observations indicate the need for models that would enable the discovery of additional novel dependencies in MLL-r B-ALLs harboring activating RAS mutations that can be exploited with combination therapy with MEK inhibitors.
Here, we report the generation of an aggressive B-ALL by the retroviral expression of mutant N-Ras in a murine knock-in Mll-Af4 mouse model. The combination of Mll-Af4 and N-Ras mutations generates an aggressive pro–B-ALL with a short latency that is serially transplantable. We found that, although the inhibition of a downstream effector of the Ras pathway, Mek, was sufficient to induce an initial response and extension of survival in vivo, leukemias still ultimately progressed. We found a potent synergism between the combination therapy of Mek inhibition and Atr inhibition, both in vitro and in vivo. Our discovery of this potent therapeutic synergism was also confirmed to be effective in patient-derived xenograft (PDX) models of human patient B-ALL samples harboring both mutations, especially those with clonal mutant RAS mutations, thus providing further evidence for this potential combination as a therapeutic approach for MLL-r B-ALL and the utility of our model as a useful preclinical platform for the discovery of insights into disease biology, as well as novel therapies in the treatment of this disease.
Methods
Generation of Mll-Af4/N-RasG12D mouse model
We previously described the generation of Mll-Af4 knock-in mice.4 Double-positive c-Kit+ and Sca1+ lineage-depleted cells from mice 6 to 8 weeks of age were sorted and grown briefly in Iscove modified Dulbecco medium containing 15% fetal bovine serum with mouse IL-7, mouse SC7, and mouse FL at 20 ng/μL (PeproTech) before retroviral transduction with virus containing MSCV-cre-IRES-TdTomato. tdTomato+ cells were sorted and transduced with MSCV-IRES-GFP (MIG) or vector containing N-RasG12D. Primary leukemias were generated by intravenous tail vein injection of 250 000 preleukemic Mll-Af4/N-RasG12D cells into lethally irradiated recipients (900 rad) with wild-type helper cells. Secondary and tertiary mice were generated by the injection of primary or secondary leukemic cells into sublethally (600 rad) irradiated recipient mice.
RNA sequencing and human gene expression analysis
Total RNA was isolated from primary leukemias and normal pro-B cells (Lin−B220+CD43+IgM−) from wild-type B6.129 mice, and libraries for RNA sequencing were prepared with a TruSeq kit (Illumina). Gene expression data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE89560. Pediatric B-ALL patient microarray expression data were obtained from GSE79450, GSE19475, and GSE77416.12,16,17 Published gene expression data from Affymetrix HG U133 Plus 2.0 arrays were processed with frozen robust multiarray analysis, a variant of robust multiarray average normalization.18
In vitro treatment experiments, cell cycle determination, and apoptosis
In vitro treatment experiments to determine 50% inhibitory concentration (IC50) with AZ20 (MedKoo; DC Chemicals), trametinib (LC Laboratories), and PD901 (LC Laboratories) were performed on cells plated in a 96-well plate. Cells were plated in triplicate and incubated with AZ20 for 72 hours. Viability was measured by DNA staining with 4′,6-diamidino-2-phenylindole and measured by flow cytometry. IC50 was calculated using GraphPad Prism. Synergism was determined by applying the Chou-Talalay combination index (CI) method.19
Immunoblotting
Primary antibodies used in these studies include phospho-Erk (T202/Y204), phospho-Chk1 (S345), phospho-Cdk1 (Y15), total-Erk, Chk1, Cdk1, GAPDH (CST), phospho-Cdk2 (Y15), total Cdk2, and phospho-γH2AX (S139) (Abcam).
In vivo treatment of animals
For all in vivo treatment experiments, 1000 secondary leukemia cells were transplanted in sublethally irradiated (600 rad) B6.129 mice through intravenous tail vein injections. Trametinib and AZ20 were administered by oral gavage, once daily. Trametinib was dissolved in 0.5% methylcellulose + 0.2% Tween-80, and AZ20 was dissolved in 10% 1-methyl-2-pyrrolidinone and 90% PEG-300. Mice were monitored daily for clinical symptoms and euthanized when they appeared moribund. All mouse experiments were approved by the Institutional Animal Care and Use Committees at Dana-Farber Cancer Institute and Memorial Sloan Kettering Cancer Center.
PDX studies
PDXs from patient samples with MLL-AF4 translocation and N-RAS mutation were obtained and transplanted in nonirradiated NSG mice (Taconic) through tail vein injections. Ten days after the injection, mice were randomized into 4 groups: vehicle, trametinib 1 mg/kg, AZ20 50 mg/kg, or combination. Mice were treated once daily by oral gavage for 15 days, and all mice were euthanized at the end of the treatment course.
Results
Retroviral expression of mutant N-RasG12D cooperates with Mll-Af4 to generate an aggressive pro–B-ALL
We previously published a murine model of Mll-Af4, in which we knocked in human AF4 into 1 allele of murine MLL with a STOP element flanking the 5′ region of the knock-in sequence. The excision of the STOP element (by Cre recombinase) results in the production of the fusion protein.4 We isolated hematopoietic stem cells from independent Mll-Af4 donor mice and introduced Cre recombinase tagged with a fluorescent tdTomato marker via retroviral transduction. These Mll-Af4–expressing cells can be propagated indefinitely in vitro in the presence of lymphoid cytokines. After the enforced expression of Cre, we transduced these transformed Mll-Af4 cells with a GFP-tagged mutant N-Ras retrovirus harboring the glycine-to-aspartic acid–activating mutation at amino acid position 12 (G12D) (Figure 1A). The retroviral expression of mutant N-RasG12D in Mll-Af4 cells rendered the transformed cells cytokine independent. Injection of double-positive cells into lethally irradiated recipients resulted in the development of leukemia, the only disease observed with this model, with a median latency of 35 days (Figure 1B). Mll-Af4/N-RasG12D leukemias were similarly aggressive in vivo upon serial transplantation, with secondary and tertiary leukemia developing at a median of 20 and 12 days after transplant into sublethally irradiated recipients, respectively (Figure 1B). The leukemic mice presented with hind leg paralysis, and tumor immunophenotyping of leukemic bone marrow indicated cell surface markers closely resembling a pro–B-cell stage of development (B220+Cd43+IgM−Cd19+Cd24+Flt3+cKit+Cd25−IgD−) (Figure 1C; supplemental Figures 1 and 2). Histological analysis revealed significant infiltration of leukemic blast cells in the bone marrow, as well as the spleen, liver, and central nervous system, a feature previously described in a mutant RAS MLL-AF4 ALL xenograft mouse model15 (Figure 1D). Sick mice displayed significant splenomegaly (Figure 1E). Polymerase chain reaction of the genomic DNA from preleukemic cells and leukemic cells confirmed that DJH recombination had occurred, indicating B-cell lineage commitment (Figure 1F).
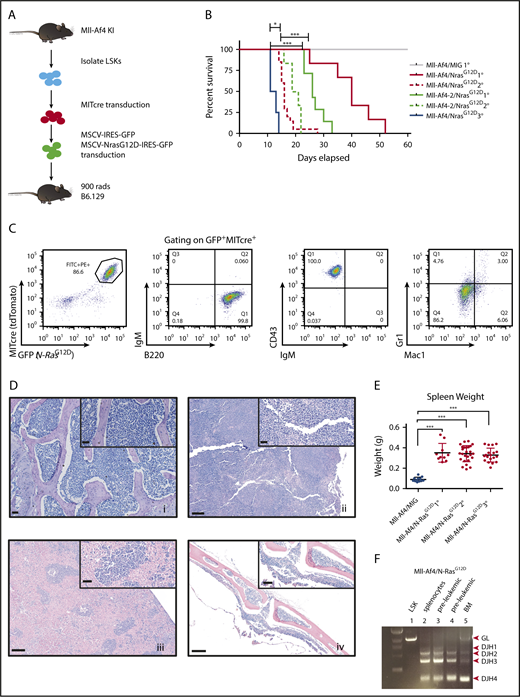
Mll-Af4 and mutant N-RasG12Dcooperate to generate an aggressive and serially transplantable B-ALL. (A) Strategy for generation of cells harboring both Mll-Af4 and mutant N-Ras by retroviral transduction. (B) Kaplan-Meier survival curves of primary, secondary, and tertiary leukemias. LSKs from 2 independent Mll-Af4 donor mice were transformed (n ≥ 5 for each donor). Mll-Af4/MIG controls did not develop disease 160 to 200 days after transplant when mice were euthanized and assessed for leukemic involvement with ≤5% detectable leukemic cells in peripheral blood or bone marrow at time of euthanization (data not shown). Secondary and tertiary leukemias were generated from injection of 100 000 primary leukemia cells (n ≥ 4 for all groups). (C) Pro-B cell (B220+Cd43+IgM−) immunophenotype of primary leukemias. (D) Histology (hematoxylin and eosin stain) of bone marrow (i), spleen (ii), liver (iii), and central nervous system (iv) from primary B-ALLs (scale bars, 200 μm). Insets show higher-magnification views (scale bars, 50 μm). (E) Spleen weights in primary (n = 10), secondary (n = 26), and tertiary (n = 19) Mll-Af4/N-RasG12D leukemic mice from ≥3 independent transplant experiments generated from 3 independent Mll-Af4 donors. Spleens from Mll-Af4/MIG (n = 12) generated from 3 independent donor mice were harvested at 160 to 200 days posttransplant. (F) Polymerase chain reaction detection of immunoglobulin rearrangement in genomic DNA from Mll-Af4/N-RasG12D preleukemic cells and in the spleen and bone marrow of primary leukemias. ***P < .001, *P < .05.
Mll-Af4 and mutant N-RasG12Dcooperate to generate an aggressive and serially transplantable B-ALL. (A) Strategy for generation of cells harboring both Mll-Af4 and mutant N-Ras by retroviral transduction. (B) Kaplan-Meier survival curves of primary, secondary, and tertiary leukemias. LSKs from 2 independent Mll-Af4 donor mice were transformed (n ≥ 5 for each donor). Mll-Af4/MIG controls did not develop disease 160 to 200 days after transplant when mice were euthanized and assessed for leukemic involvement with ≤5% detectable leukemic cells in peripheral blood or bone marrow at time of euthanization (data not shown). Secondary and tertiary leukemias were generated from injection of 100 000 primary leukemia cells (n ≥ 4 for all groups). (C) Pro-B cell (B220+Cd43+IgM−) immunophenotype of primary leukemias. (D) Histology (hematoxylin and eosin stain) of bone marrow (i), spleen (ii), liver (iii), and central nervous system (iv) from primary B-ALLs (scale bars, 200 μm). Insets show higher-magnification views (scale bars, 50 μm). (E) Spleen weights in primary (n = 10), secondary (n = 26), and tertiary (n = 19) Mll-Af4/N-RasG12D leukemic mice from ≥3 independent transplant experiments generated from 3 independent Mll-Af4 donors. Spleens from Mll-Af4/MIG (n = 12) generated from 3 independent donor mice were harvested at 160 to 200 days posttransplant. (F) Polymerase chain reaction detection of immunoglobulin rearrangement in genomic DNA from Mll-Af4/N-RasG12D preleukemic cells and in the spleen and bone marrow of primary leukemias. ***P < .001, *P < .05.
We conducted gene set enrichment analysis of RNA sequencing of primary Mll-Af4/N-RasG12D B-ALLs, sorted normal pro-B cells (Lin−B220+CD43+IgM−), and found significant correlation with a signature consistent with glucocorticoid-persistent B-ALLs (supplemental Figure 3A),20 an insensitivity often observed in MLL-AF4+ B-ALLs.1,10,13,14 Unexpectedly, our Mll-Af4/N-RasG12D B-ALLs did not have high Hoxa cluster or Meis1 expression (supplemental Figure 3B). However, low HOXA–expressing human MLL-AF4+ leukemia is a distinct subgroup within MLL-AF4 B-ALLs with higher risk for relapse.12 Furthermore, activating RAS mutations have been found to be co-occurring with low HOXA–expressing MLL-AF4+ B-ALLs,21 further indicating the utility and biological relevance of our GEMM in modeling this disease. Together, these data suggest that the coexpression of activated N-Ras and Mll-Af4 is necessary to generate a highly aggressive serially transplantable B-ALL in vivo.
Inhibition of mutant Ras signaling is insufficient as a single agent to prevent disease progression
Because the expression of mutant N-RasG12D was sufficient to allow Mll-Af4–expressing cells to be cytokine independent in vitro, we hypothesized that these leukemic cells would be particularly dependent on the downstream effectors of N-Ras, such as Mek. MEK inhibition of MLL-r infant ALLs harboring mutant RAS has been reported to induce apoptosis in vitro.13-15 To test the dependency of these transformed cells on the N-Ras/Mek signaling pathway, we treated preleukemic Mll-Af4/N-RasG12D cells and empty vector control (Mll-Af4/MIG) cells with 2 MEK inhibitors: trametinib (GSK212) and PD901. Mll-Af4/N-RasG12D preleukemic cells grown in the absence of cytokines displayed increased sensitivity to Mek inhibition (IC50 ∼ 2 nM) compared with those grown in normal lymphoid cytokine conditions (IC50 ∼ 5 nM) or transduced with an empty vector control (IC50 ∼ 10 nM) (Figure 2A). Similar IC50 values were found when cells were treated with PD901 (data not shown). We chose to use trametinib for subsequent studies because it had previously been shown to have good in vivo biological activity and is well tolerated, more specifically, in leukemia models harboring N-Ras mutations.22,23 We were able to confirm the inhibition of the Mek pathway in Mll-Af4/N-RasG12D preleukemic cells with trametinib by the dose-dependent reduction of phospho-Erk (Figure 2B) upon treatment. We next asked whether we could observe a similar response in vivo. We transplanted secondary Mll-Af4/N-RasG12D leukemias into sublethally irradiated recipient mice (Figure 2C). Mice were treated, after detectable engraftment of blasts in the peripheral blood, with vehicle or with trametinib (0.5 mg/kg or 1 mg/kg) by oral gavage once daily for 7 days and then analyzed for leukemic involvement. Mll-Af4/N-RasG12D mice treated with trametinib exhibited dose-dependent responses in the reduction of leukemia cells in the bone marrow, spleen weight, and white blood count (Figure 2C). Additionally, phospho-Erk levels were reduced in sorted leukemic cells from treated mice at both dose levels (Figure 2D).

In vitro and in vivo sensitivity of Mll-Af4/N-RasG12DB-ALLs to MEK inhibition. (A) Mll-Af4/N-RasG12D and Mll-Af4/MIG preleukemic cells were treated with trametinib for 72 hours, and cell viability was measured by 4′,6-diamidino-2-phenylindole staining by flow cytometry. All experiments were conducted in triplicate, and data are represented as a percentage of dimethyl sulfoxide controls. (B) Phospho-Erk and total Erk protein levels of Mll-Af4/N-RasG12D mice treated with trametinib for 24 hours. (C) Leukemic burden in bone marrow, as measured by double-positive cells, white blood cell counts, and spleen weights of mice injected with 1000 secondary Mll-Af4/N-RasG12D leukemic cells and treated in vivo once daily for 7 days, by oral gavage, with vehicle or trametinib (0.5 mg/kg or 1 mg/kg) 14 days after injection (red arrows). (D) Phospho-Erk levels in leukemic bone marrow cells (sorted for tdTomato and GFP double positivity) in mice treated for 7 days with trametinib by oral gavage or vehicle controls. (E) Kaplan-Meier survival plot of prolonged treatment with trametinib. (F) Phospho-Erk levels of leukemic cells from the bone marrow of mice on short-term (ST; 7 days), long-term (LT; ≥20 days), or prolonged treatment with trametinib (1 mg/kg) or from vehicle controls (from Figure 1E). For all in vivo experiments, n ≥ 4 mice per group. Data are representative of ≥3 independent treatment experiments. ***P < .001, **P < .01.
In vitro and in vivo sensitivity of Mll-Af4/N-RasG12DB-ALLs to MEK inhibition. (A) Mll-Af4/N-RasG12D and Mll-Af4/MIG preleukemic cells were treated with trametinib for 72 hours, and cell viability was measured by 4′,6-diamidino-2-phenylindole staining by flow cytometry. All experiments were conducted in triplicate, and data are represented as a percentage of dimethyl sulfoxide controls. (B) Phospho-Erk and total Erk protein levels of Mll-Af4/N-RasG12D mice treated with trametinib for 24 hours. (C) Leukemic burden in bone marrow, as measured by double-positive cells, white blood cell counts, and spleen weights of mice injected with 1000 secondary Mll-Af4/N-RasG12D leukemic cells and treated in vivo once daily for 7 days, by oral gavage, with vehicle or trametinib (0.5 mg/kg or 1 mg/kg) 14 days after injection (red arrows). (D) Phospho-Erk levels in leukemic bone marrow cells (sorted for tdTomato and GFP double positivity) in mice treated for 7 days with trametinib by oral gavage or vehicle controls. (E) Kaplan-Meier survival plot of prolonged treatment with trametinib. (F) Phospho-Erk levels of leukemic cells from the bone marrow of mice on short-term (ST; 7 days), long-term (LT; ≥20 days), or prolonged treatment with trametinib (1 mg/kg) or from vehicle controls (from Figure 1E). For all in vivo experiments, n ≥ 4 mice per group. Data are representative of ≥3 independent treatment experiments. ***P < .001, **P < .01.
Because our transplanted mice seemed to tolerate trametinib treatment, we next determined whether we could sustain long-term durable disease-free mice upon prolonged treatment. We transplanted secondary Mll-Af4/N-RasG12D leukemias and initiated treatment 7 days after injection. Treatment was continued until the mice appeared moribund or exhibited signs of disease. Although we were able to significantly increase survival of mice with trametinib, the treated mice ultimately developed disease at both drug doses (Figure 2E; supplemental Figure 4). Additionally, the mice that had undergone prolonged treatment with trametinib exhibited a loss of sensitivity to Mek inhibition, as measured by the presence of phospho-Erk in leukemic bone marrow cells at the time of euthanization (Figure 2F). These data indicate that trametinib alone can prolong survival in this model but is insufficient to prevent leukemic progression and is consistent with findings of another recent study using trametinib as monotherapy against MLL-r infant B-ALL with activated RAS.15
Mll-Af4/N-RasG12D B-ALL cells are sensitive to ATR inhibition
Because single-agent Mek inhibition was insufficient to generate long-term durable responses, we looked at our gene set enrichment analysis of primary Mll-Af4/NrasG12D B-ALLs to determine additional pathways of interest that could be therapeutically tractable. Interestingly, our analysis revealed significant negative correlations with gene signatures involving the G2/M checkpoint and ATR activation in response to replication stress (supplemental Figure 5), suggesting derangements in the replication stress response (RSR) and DNA damage response (DDR) pathways.
Several recent studies have demonstrated that high levels of oncogene-driven replicative stress in MLL-r acute myelogenous leukemia (AML) render them particularly dependent on the master regulator of intra-S phase and G2/M cell cycle checkpoint, ATR kinase.24-28 Additionally, there has been evidence that ATR directly phosphorylates MLL, and this event is required for the G2-M checkpoint.29 Inhibition of ATR has been shown to induce p53-independent apoptosis30-34 and have a strong antitumoral effect in vivo.25,35 Inhibitors of ATR and CHK1 kinase, the major phosphorylation target of ATR, have shown efficacy in multiple hematopoietic tumor types and are being assessed in preclinical and clinical studies, especially in malignancies with deficiencies in DDR/RSR components.27,28,36-46
To determine whether the Atr signature downregulation that we observed in our mouse model was also a feature of B-ALL in human disease, we analyzed expression data from several published data sets comparing MLL-r B-ALLs with normal pre-B controls.12,16,17 We found that a large majority of human MLL-AF4 infant leukemias exhibited significant signature-wide downregulation of the expression of genes in the ATR signature compared with normal pre-B cells, as well as with myeloid hematopoietic stem and progenitor cell controls (supplemental Figure 6). These data suggest that our mouse model faithfully recapitulates features of infant B-ALL harboring MLL-AF4 translocations, as well as points to targeting the DDR/RSR and ATR pathways as a potential novel therapeutic strategy.
Because our model contains an Mll fusion protein, as well as an activated signaling protein, N-RasG12D, we expected higher levels of DNA damage due to elevated oncogene-induced replicative stress. To confirm this, we measured the basal levels of DNA damage by assessing the phospho-γH2Ax levels in our Mll-Af4/N-RasG12D–driven leukemias. By immunofluorescence (supplemental Figure 7A), we were able to detect higher basal levels of phospho-γH2AX in Mll-Af4/N-RasG12D–expressing preleukemic cells.
Having observed increased replication stress and altered expression of Atr pathway members in our model of Mll-r B-ALL, we hypothesized that, if we could further attenuate the RSR/DDR response by further inhibiting Atr in these leukemias, we could potentially exploit the dependency of these leukemic cells on this pathway and, thereby, enhance the induction of leukemic cytotoxicity. To test this hypothesis, we targeted Atr with AZ20, which has previously been shown to have bioactivity in vivo.35 In vitro treatment with AZ20 showed antiproliferative effects in Mll-Af4 and Mll-Af4/N-RasG12D preleukemic cells at submicromolar concentrations (Figure 3A), which were lower than Mll-Af9 primary leukemias that were generated by retroviral transduction of LSKs (retro)47 or from knock-in Mll-Af9 mice (KI)48 and previously shown to be sensitive to AZ20 treatment.25 Additionally, the sensitivity of our mouse models of B-ALL and AML appeared to be correlated with their in vitro proliferation rate (Figure 3B). Inhibition of Atr with AZ20 was sufficient to reduce the phosphorylation and activation of its direct target, Chk1, and abrogate the G2/M checkpoint through a Cdk1-dependent mechanism (Figure 3C-D), which had been previously reported in MLL-r AMLs.34 Furthermore, inhibition of ATR induced DNA damage, as measured by the increase in phospho-γH2Ax levels (supplemental Figure 7B) and apoptosis (Figure 3E) in a dose-dependent manner. Moreover, the p53 pathway was intact in Mll-r B-ALLs, because treatment with the MDM2 antagonist Nutlin-3 could efficiently induce apoptosis (supplemental Figure 7C-D). Based on these in vitro results, we were curious to see whether Atr inhibition, as a single agent, could induce leukemic cell death in vivo. Although once-a-day treatment with AZ20 extended the survival of mice injected with highly aggressive secondary Mll-Af4/N-RasG12D leukemia cells, they ultimately succumbed to disease (Figure 3F). This indicates that, even though Atr inhibition alone did have transient efficacy in this model, it was insufficient for durably inhibiting disease progression.

Sensitivity of Mll-Af4/N-RasG12DB-ALLs to Atr inhibition. (A) IC50 summary of 72-hour treatment of Mll-Af4 B-ALLs vs Mll-Af9 AMLs. Mll-Af4/N-RasG12D preleukemic cells, Mll-Af9 primary leukemias from retroviral models (Retro), and Mll-Af9 knock-in mice (KI) were treated with AZ20, as previously described. One-way ANOVA analyses were conducted to calculate multivariate significance between MLL-r B-ALLs and MLL-r AMLs. (B) Correlation of mouse model line IC50 values treated with AZ20 and fold expansion of cell lines at 72 hours. Fold expansion is expressed as a ratio of total cell count in dimethyl sulfoxide (DMSO) controls to initial plating. (C) Phospho- and total Chk1 (S345), Cdk1 (Y15), and Cdk2 (Y15) immunoblotting of Mll-Af4/N-RasG12D preleukemic cells treated for 24 hours with 0, 1, or 3 μM AZ20. (D) Representative cell cycle plots for representative primary murine Mll-Af9 AMLs generated by retroviral transformation (#45) and preleukemic Mll-Af4/NrasG12D B-ALLs treated for 24 hours with DMSO and 3 μM AZ20. The plots are representative of ≥3 independent experiments. (E) Annexin V+Mll-Af9 AMLs and Mll-Af4 B-ALLs were treated for 24 hours with 0, 1, or 3 μM AZ20. (F) Treatment of secondary leukemias with 50 mg/kg AZ20 alone, 7 days after transplant (red arrow), extended the survival of treated mice. All in vitro data are representative of n ≥ 5 independent treatment experiments. For in vivo treatment experiments, n ≥ 4 mice per group were used. ***P < .001, **P < .01, *P < .05.
Sensitivity of Mll-Af4/N-RasG12DB-ALLs to Atr inhibition. (A) IC50 summary of 72-hour treatment of Mll-Af4 B-ALLs vs Mll-Af9 AMLs. Mll-Af4/N-RasG12D preleukemic cells, Mll-Af9 primary leukemias from retroviral models (Retro), and Mll-Af9 knock-in mice (KI) were treated with AZ20, as previously described. One-way ANOVA analyses were conducted to calculate multivariate significance between MLL-r B-ALLs and MLL-r AMLs. (B) Correlation of mouse model line IC50 values treated with AZ20 and fold expansion of cell lines at 72 hours. Fold expansion is expressed as a ratio of total cell count in dimethyl sulfoxide (DMSO) controls to initial plating. (C) Phospho- and total Chk1 (S345), Cdk1 (Y15), and Cdk2 (Y15) immunoblotting of Mll-Af4/N-RasG12D preleukemic cells treated for 24 hours with 0, 1, or 3 μM AZ20. (D) Representative cell cycle plots for representative primary murine Mll-Af9 AMLs generated by retroviral transformation (#45) and preleukemic Mll-Af4/NrasG12D B-ALLs treated for 24 hours with DMSO and 3 μM AZ20. The plots are representative of ≥3 independent experiments. (E) Annexin V+Mll-Af9 AMLs and Mll-Af4 B-ALLs were treated for 24 hours with 0, 1, or 3 μM AZ20. (F) Treatment of secondary leukemias with 50 mg/kg AZ20 alone, 7 days after transplant (red arrow), extended the survival of treated mice. All in vitro data are representative of n ≥ 5 independent treatment experiments. For in vivo treatment experiments, n ≥ 4 mice per group were used. ***P < .001, **P < .01, *P < .05.
Dual inhibition of Atr and Mek enhances Mll-Af4/N-RasG12D leukemic cell cytotoxicity in vitro
Because single-agent MEK or ATR inhibition had some cytotoxic effect on leukemic cells but alone could not prevent leukemic progression in vivo, we wanted to ascertain whether there would be an effect of the combination of dual ATR and MEK inhibition. We treated Mll-Af4/NrasG12D preleukemic cells in vitro with combinations of AZ20 (0-3 μM) and trametinib (0-33 nM) for 72 hours and assessed viability. Synergy was evaluated by calculation of a CI.19 Combinations that fall below the line of additivity are considered synergistic. For all combinations, the CI values fell below the line of additivity (CI < 1) (Figure 4A), indicating strong synergy. Furthermore, in vitro experiments showed that, in combination, concentrations of trametinib and AZ20 at or well below their single-agent IC50 value demonstrated significant antiproliferative and apoptotic effects on Mll-Af4/N-RasG12D preleukemic cells (Figure 4B-C).

Synergistic in vitro antileukemic effects of dual inhibition of MEK and ATR in Mll-Af4/N-RasG12DB-ALL. (A) Normalized isobologram for the nonconstant ratio combination design (Chou-Chou plot) of in vitro drug combinations. Mll-Af4/NrasG12D preleukemic cells were treated with combinations of AZ20 and trametinib (GSK212) for 72 hours at doses ranging from 0 to 3 μM AZ20 and from 0 to 33 nM GSK212. Synergy was evaluated using the Chou-Talalay CI for Loewe additivity in R. The data are representative of n > 3 independent combination treatment experiments. (B) Annexin V+Mll-Af4 B-ALLs were treated for 24 hours with single-agent GSK212 (1 nM) or AZ20 (0.5 μM) or with the combination. One-way ANOVA analyses were conducted to calculate multivariate significance between dimethyl sulfoxide controls and single-agent treatments to combination treatment. (C) Viability of treatment of Mll-Af4/N-RasG12D preleukemic cells with GSK212 and AZ20 at concentrations well below the single-agent IC50 values and represented as a percentage of DMSO controls. All experiments were performed in triplicate. The results are shown as an average. ***P < .001.
Synergistic in vitro antileukemic effects of dual inhibition of MEK and ATR in Mll-Af4/N-RasG12DB-ALL. (A) Normalized isobologram for the nonconstant ratio combination design (Chou-Chou plot) of in vitro drug combinations. Mll-Af4/NrasG12D preleukemic cells were treated with combinations of AZ20 and trametinib (GSK212) for 72 hours at doses ranging from 0 to 3 μM AZ20 and from 0 to 33 nM GSK212. Synergy was evaluated using the Chou-Talalay CI for Loewe additivity in R. The data are representative of n > 3 independent combination treatment experiments. (B) Annexin V+Mll-Af4 B-ALLs were treated for 24 hours with single-agent GSK212 (1 nM) or AZ20 (0.5 μM) or with the combination. One-way ANOVA analyses were conducted to calculate multivariate significance between dimethyl sulfoxide controls and single-agent treatments to combination treatment. (C) Viability of treatment of Mll-Af4/N-RasG12D preleukemic cells with GSK212 and AZ20 at concentrations well below the single-agent IC50 values and represented as a percentage of DMSO controls. All experiments were performed in triplicate. The results are shown as an average. ***P < .001.
In vivo efficacy of dual inhibition of Atr and Mek in Mll-Af4/N-RasG12D B-ALL
We next wanted to see whether the efficacy that we found in our in vitro combination therapy inhibitors studies would translate in vivo. Secondary Mll-Af4/N-RasG12D leukemic cells were transplanted into sublethally irradiated recipients, which were treated by oral gavage, once daily, with vehicle, AZ20 (50 mg/kg), trametinib (0.5 mg/kg), or the combination for 14 days after leukemic cells were detected in the peripheral blood. Strikingly, mice treated with the combination had significantly delayed progression to disease (Figure 5A; supplemental Figures 8 and 9). In fact, mice treated with the combination only developed disease after the withdrawal of drug after an already-significant extension of survival over mice treated with single-agent inhibitor (Figure 5A). In all hematopoietic tissues assessed, we observed significant reductions in leukemia burden (Figure 5B; supplemental Figures 8 and 9). Furthermore, we observed increased apoptosis in mice treated with the combination over single-agent– or vehicle-treated mice and a dramatic reduction in leukemic infiltration in all hematopoietic tissues assessed (Figure 5C; supplemental Figures 8-10). Additionally, phospho-Erk was undetectable in sorted combination-treated leukemic cells, in sharp contrast to what had been observed in cells from mice after long-term single-agent treatment. This suggests that the combination of trametinib and AZ20 can maintain the sustainable inhibition of activated Mek in a model with activating mutations in N-Ras (Figures 2F and 5D).

In vivo antileukemic efficacy of dual inhibition of MEK and ATR in Mll-Af4/N-RasG12DB-ALL. (A) Kaplan-Meier survival plot of mice transplanted with 1000 secondary B-ALL cells and treated with vehicle, single-agent trametinib (0.5 mg/kg), single-agent AZ20 (50 mg/kg), or the combination. Treatment of mice began 7 days posttransplant (single red arrow) and was sustained throughout the experiment. In the combination-treatment cohort, treatment was suspended on day 32 posttransplant (double red arrows). (B) Leukemia burden, measured by double positivity of GFP and tdTomato, was assessed in several hematopoietic tissues of mice treated for 14 days after detectable engraftment of leukemic cells in the peripheral blood. (C) Apoptosis, as measured by Annexin V+, in double-positive leukemia cells of mice treated with trametinib, AZ20, or the combination. (D) Phospho-Erk levels in sorted leukemia cells from mice treated with a combination of MEK and ATR inhibitors. The data are representative of 3 independent treatment experiments with 2 different leukemias derived from independent donors (supplemental Figures 8 and 9). ***P < .001, **P < .01, *P < .05.
In vivo antileukemic efficacy of dual inhibition of MEK and ATR in Mll-Af4/N-RasG12DB-ALL. (A) Kaplan-Meier survival plot of mice transplanted with 1000 secondary B-ALL cells and treated with vehicle, single-agent trametinib (0.5 mg/kg), single-agent AZ20 (50 mg/kg), or the combination. Treatment of mice began 7 days posttransplant (single red arrow) and was sustained throughout the experiment. In the combination-treatment cohort, treatment was suspended on day 32 posttransplant (double red arrows). (B) Leukemia burden, measured by double positivity of GFP and tdTomato, was assessed in several hematopoietic tissues of mice treated for 14 days after detectable engraftment of leukemic cells in the peripheral blood. (C) Apoptosis, as measured by Annexin V+, in double-positive leukemia cells of mice treated with trametinib, AZ20, or the combination. (D) Phospho-Erk levels in sorted leukemia cells from mice treated with a combination of MEK and ATR inhibitors. The data are representative of 3 independent treatment experiments with 2 different leukemias derived from independent donors (supplemental Figures 8 and 9). ***P < .001, **P < .01, *P < .05.
Dual inhibition of ATR and MEK is effective in PDX models of MLL-AF4 B-ALL harboring activated N-RAS mutations
Having observed in vivo efficacy of Atr and Mek combination therapy in our mouse model of B-ALL, we wanted to determine whether these results would be translatable to human B-ALL patient samples harboring both MLL-AF4 and activating N-RAS mutations. To this end, we obtained patient samples that had MLL-AF4 rearrangement and activating N-RAS mutations (supplemental Table 1) and tested the response of these leukemic cells in PDX models. The transplanted mice were treated with vehicle, single agent, or the combination for 14 days before all mice were euthanized and analyzed for leukemic involvement in the blood, bone marrow, and spleen. As we had seen in our mouse model of B-ALL, mice treated with the combination of MEK and ATR inhibitors showed a significant reduction in leukemic involvement in all hematopoietic tissues assessed (Figure 6). The combination was more potent in the PDX with higher initial mutant NRAS variant allele frequency (VAF) (PDX 83; NRAS G13D VAF ∼ 50%) compared with the PDX with lower initial VAF (PDX 24; NRAS Q61R VAF ∼ 18%, G12S VAF ∼ 2%) (Figure 6; supplemental Figure 11; supplemental Table 1), likely due to the elimination of the mutant RAS–containing subclones in that sample (supplemental Figure 11). These data support further preclinical development of the combination of trametinib and AZ20 for the potential treatment of MLL-AF4/N-RAS B-ALLs, particularly those with high mutant RAS VAF.

Antileukemic efficacy of combination MEK and ATR inhibition in human MLL-AF4 mutant N-RAS B-ALL in PDX models. Patient samples harboring MLL-AF4 and mutant N-Ras were injected in NSG mice that were treated with vehicle, single-agent trametinib (1 mg/kg), single-agent AZ20 (50 mg/kg), or the combination for 15 days. Percentage of human leukemic blasts in hematopoietic tissues and spleen weights of treatment cohorts for PDX 83 (A) and PDX 24 (B). For all vehicle and single-agent treatment groups, n ≥ 4 mice; for the combination group, n = 3. ***P < .001, **P < .01, *P < .05.
Antileukemic efficacy of combination MEK and ATR inhibition in human MLL-AF4 mutant N-RAS B-ALL in PDX models. Patient samples harboring MLL-AF4 and mutant N-Ras were injected in NSG mice that were treated with vehicle, single-agent trametinib (1 mg/kg), single-agent AZ20 (50 mg/kg), or the combination for 15 days. Percentage of human leukemic blasts in hematopoietic tissues and spleen weights of treatment cohorts for PDX 83 (A) and PDX 24 (B). For all vehicle and single-agent treatment groups, n ≥ 4 mice; for the combination group, n = 3. ***P < .001, **P < .01, *P < .05.
Discussion
Although genomic approaches have informed the biology of MLL-r ALL, we still lack highly effective treatments for this disease. A complicating factor that has slowed progress in this area is the absence of accurate murine and human models that recapitulate the genetic and clinical features of this disease. The inability to generate aggressive disease with the expression of MLL-AF4 alone indicates that our understanding of the cellular origins and/or transformation potential of this disease is incomplete. Recent next-generation deep sequencing of relapse samples from MLL-AF4 patients have indicated that Ras mutations are present at a high frequency (∼69%) in paired samples at diagnosis, indicating that subclones harboring RAS mutations were present at diagnosis and, furthermore, are of therapeutic interest because they survive initial therapy.16 Another recent targeted deep sequencing study of infant ALLs further confirmed that RAS mutations are present at a high frequency in MLL-r patients (∼73%), with ∼40% of these having activating RAS mutations with VAF > 10%.13 Furthermore, B-ALLs harboring RAS mutations seem to confer resistance to glucocorticoids and standard chemotherapeutic agents.10,13,14 Given the increasing body of evidence suggesting that inhibiting downstream effectors of RAS alone is insufficient in the treatment of this disease, we sought to generate a mouse model that would allow for a more detailed interrogation of the biology of this subtype of B-ALL and would lead to the discovery of additional dependencies that could be exploited in combination therapies. Here, we demonstrate that the combination of Mll-Af4 and activating N-Ras mutations is sufficient to transform bone marrow cells and render them cytokine independent in vitro, as well as to give rise to an extremely aggressive B-ALL that is serially transplantable in vivo.
Although single-agent Mek or Atr inhibition alone was insufficient to prevent leukemic progression, the combination seemed to be potent in its ability to induce leukemic cell cytotoxicity. Furthermore, the combination was effective in repressing the reactivation of the Mek pathway that was observed following trametinib single-agent treatment. As further evidence that this combination could be useful for patients with this disease, we tested the combination on primary B-ALL patient samples harboring MLL-AF4 rearrangement and N-RAS mutation in PDX models and demonstrated significant antileukemic effects, particularly in samples with high mutant RAS VAFs.
The acute response to ATR inhibitors in our mouse model harboring activating Ras mutations is in line with previous data indicating that activating Ras mutations exacerbate synthetic lethality with ATR inhibition.49 However, the derangements of the Atr pathway observed in our mouse models of B-ALL were more unexpected. Although the signaling components of the Atr/DDR pathway are regulated at the protein level through protein phosphorylation, the response is also dependent on the expression of other DNA damage complex members (eg, Timeless, Tipin, Topbp1, Clspn, Rad, and Rfc family genes) whose lower expression may impact the overall cellular response to perturbations in the ATR/DDR/RSR pathways. Furthermore, previous studies have suggested that, in AML, higher levels of expression of CHK1 may serve to buffer the high levels of replicative stress present in these cells.25,44 However, our data suggest that MLL-r B-ALLs exist in a precarious steady-state with increased replicative stress and downregulation of Atr pathway members. Together, these features may render these cells highly susceptible to further perturbations of the RSR pathway. Additionally, our data suggest that MLL-r B-ALLs are highly sensitive to ATR inhibition that is due, in part, to the increased proliferative rate of this acute leukemia. Thus, in this context, ATR inhibition can be used as a cytostatic and cytotoxic agent in these highly proliferative acute leukemia cells, potentially providing an alternative to the use of, and obviating the need for, chemotherapy in combination-therapy strategies. The observation that a majority of MLL-AF4 patient samples also exhibited signature-wide downregulation of the ATR pathway suggests that, in these patients, further inhibition of the DDR/ATR pathways to sensitize or provide synthetic lethality, in combination with other targeted therapies, could be therapeutically effective (eg, combinations with glucocorticoids and BET50 inhibitors), especially for patients with subclonal RAS mutations.
Altogether, our model of B-ALL recapitulates features of human disease and can be used as a platform for the discovery of novel therapeutic combinations, as well as for studying the underlying molecular and genetic mechanisms driving leukemogenesis.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE89560).
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Armstrong Laboratory for helpful feedback and fruitful scientific suggestions. They also thank Servicebio Inc. for performing histological analyses for these studies.
This work was supported by National Institutes of Health, National Cancer Institute grants R01 CA176745 and 5P01 CA066996 (S.A.A.). S.H.C. is a Damon Runyon-Sohn Pediatric Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-5-13). E.J.S. is supported by the Howard Hughes Medical Institute Medical Research Fellows Program and was a Howard Hughes Medical Institute Research Fellow. The Nussenzweig Laboratory is supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, the Center for Cancer Research, and an Alex’s Lemonade Stand Foundation Award.
Authorship
Contribution: S.H.C. designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript; E.J.S. designed and performed experiments and analyzed and interpreted the data; J.M., J.R.C., and C.N.M. performed experiments and analyzed and interpreted the data; R.P.K., K.R., J.L.M., and E.S.F. provided computational analysis for RNA sequencing, expression data, and VAF analyses; A.K. provided the pediatric patient samples, A.N. and H.X. provided reagents, Z.F. provided logistical support; and S.A.A. helped to design experiments, interpreted the results, supervised the study, and helped with manuscript preparation.
Conflict-of-interest disclosure: S.A.A. consults for Epizyme and is on the scientific advisory board for Imago Biosciences, C4 Therapeutics, and Cyteir Therapeutics and received research funding from AstraZeneca. The remaining authors declare no competing financial interests.
Correspondence: Scott A. Armstrong, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA 02215-5450; e-mail: scott_armstrong@dfci.harvard.edu.

