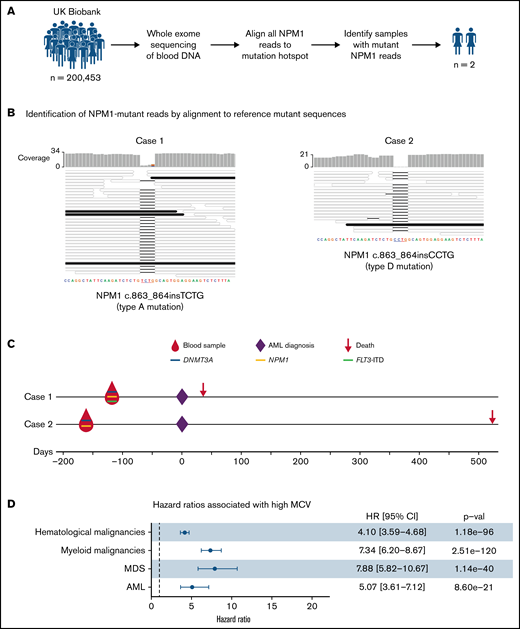
TO THE EDITOR:
Acute myeloid leukemia (AML) is an aggressive hematological malignancy characterized by effacement of normal hematopoiesis by undifferentiated myeloid blasts and failure of mature blood cell production.1 Frontline anti-AML therapy has not changed substantially for decades and, despite recent therapeutic progress, the disease remains lethal to the majority of sufferers.2-4 Furthermore, and unlike many other lethal cancers, it has not previously been considered plausible to prevent or delay the development of AML. Recent advances have revealed that AML commonly evolves from the benign phenomenon of clonal hematopoiesis (CH), the expansion of a hematopoietic stem cell and its progeny in association with leukemia-associated somatic mutations.5-8 This fate is uncommon and befalls only ∼1% of individuals with CH; however, it has been shown that cases of CH at high risk of AML can be identified years in advance,9,10 raising hopes that AML prevention may be plausible.11 The concept has received further support from preclinical reports that targeted interventions may delay or avert leukemic progression to NPM1-mutant AML, the most common AML subtype.12 However, NPM1 mutations are thought to be AML defining13 and have not been previously identified prior to the onset of myeloid malignancy,5-10,14 raising doubts about whether they can be detected in time for preventive interventions to be administered.
To investigate whether individuals with NPM1 mutations can be identified robustly and in a time and manner that could facilitate interventions to prevent AML development, we applied a bespoke approach to analyze whole exome sequencing (WES) of blood DNA from 200 453 UK Biobank (UKBB) participants, for whom detailed linked health records are available.15 In particular, we exploited the fact that over 98% of NPM1 mutations are in the form of a 4-nucleotide insertion/duplication,16-18 a change that cannot easily be generated by sequencing error. To maximize sensitivity for detecting every read reporting these mutations, we first constructed reference sequences for the 3 most common NPM1 somatic variants, namely type A (c.863_864insTCTG), type B (c.863_864insCATG), and type D (c.863_864insCCTG) (supplemental Table 1), which together represent ∼90% of all NPM1 mutations in AML.17 WES reads were aligned to the human genome assembly GRCh38 using Burrows-Wheeler Aligner Maximal Exact Match (BWA-MEM) 0.7.17.19 Reads aligned to NPM1 (chr5:171 381,174-171 416,825) were extracted with Samtools 1.920 and realigned to the constructed sequences using BWA-MEM 0.7.17.19 After realignment, reads matching any of the 3 mutation types were identified by scanning the “CIGAR string” and “optional field” of the BWA output using customized scripts. Additional myeloid gene mutations in individuals with NPM1 mutations were identified using Mutect2 (https://gatk.broadinstitute.org) and a modified version of RNAmut.21 Complete blood count data for female participants aged 55 to 65 were extracted from the UKBB. For details, also see supplemental Methods.
Our analysis identified only 2 individuals with sequencing reads reporting an NPM1 hotspot mutation: case 1, with 4 of 32 reads reporting the canonical type A and case 2, with 1 of 19 reads reporting a type D NPM1 mutation (Figure 1A-B). To search for other AML-associated somatic gene mutations in cases 1 and 2, we analyzed their blood DNA WES data using Mutect2. This identified a mutation in DNMT3A in both cases, namely DNMT3A c.2645G>A (p.Arg882His) in case 1 (variant allele fraction (VAF) = 0.24) and DNMT3A c.1627G>T (p.Gly543Cys) (VAF = 0.18) in case 2 (supplemental Table 2). Expectedly, Mutect2 only identified the NPM1 mutation in case 1 (NPM1 c.863_864insTCTG; VAF = 0.13; supplemental Table 1) as case 2 only had a single mutant read. In addition, we used a modification of the bespoke mutation detection software RNAmut21 to specifically search for internal tandem duplications in the FLT3 gene (FLT3-ITD), a mutation that commonly cooccurs with mutant NPM122 but can be missed by mutation callers.21 This identified a FLT3-ITD mutation in case 1 (VAF 0.09; supplemental Figure 1).
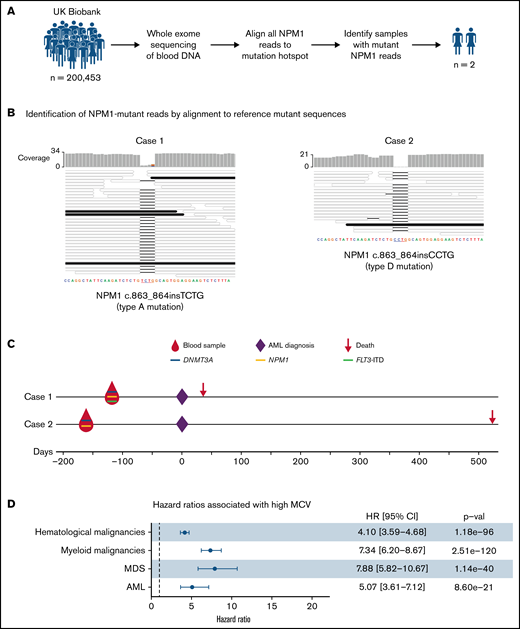
Detection and significance of NPM1 mutations in blood DNA of healthy individuals. (A) Approach used to identify NPM1 gene mutations in WES of blood DNA from 200 453 UKBB participants. (B) Alignment of sequencing reads from the 2 cases with NPM1 mutations against reference type A (left) and type D (right) NPM1 mutations. This identified 4 reads reporting the type A mutations in case 1 (left) and 1 read reporting the type D mutation in case 2 (right). Mutant reads (black horizontal bars) match perfectly with their respective reference mutant sequences, whereas wild-type reads (colorless horizontal bars) align with a 4-nucleotide gap at the insertion/duplication hotspot. (C) Timeline, gene mutations, and outcomes of the 2 individuals with NPM1 mutations. Both cases were also found to harbor mutations in the DNMT3A gene, whilst case 1 also harbored an internal tandem duplication in the FLT3 gene. (D) Forest plot of hazard ratios for hematological malignancies, myeloid malignancies, AML, and MDS associated with a high MCV (MCV > 99.5 fl) in the UKBB. CI, confidence interval; HR, hazard ratio; MCV, mean corpuscular volume; MDS, myelodysplastic syndrome.
Detection and significance of NPM1 mutations in blood DNA of healthy individuals. (A) Approach used to identify NPM1 gene mutations in WES of blood DNA from 200 453 UKBB participants. (B) Alignment of sequencing reads from the 2 cases with NPM1 mutations against reference type A (left) and type D (right) NPM1 mutations. This identified 4 reads reporting the type A mutations in case 1 (left) and 1 read reporting the type D mutation in case 2 (right). Mutant reads (black horizontal bars) match perfectly with their respective reference mutant sequences, whereas wild-type reads (colorless horizontal bars) align with a 4-nucleotide gap at the insertion/duplication hotspot. (C) Timeline, gene mutations, and outcomes of the 2 individuals with NPM1 mutations. Both cases were also found to harbor mutations in the DNMT3A gene, whilst case 1 also harbored an internal tandem duplication in the FLT3 gene. (D) Forest plot of hazard ratios for hematological malignancies, myeloid malignancies, AML, and MDS associated with a high MCV (MCV > 99.5 fl) in the UKBB. CI, confidence interval; HR, hazard ratio; MCV, mean corpuscular volume; MDS, myelodysplastic syndrome.
Strikingly, both case 1 and case 2 developed AML 133 and 168 days after blood sample donation and, unfortunately, died 36 and 536 days after diagnosis, respectively (Figure 1C). Both were previously well and had no significant past medical history. Their complete blood count results at the time of donation showed only mild abnormalities such as a raised MCV (Table 1; Figure 1D) that would not ordinarily trigger hematological investigations. Nevertheless, the short latency between detection of mutant NPM1 and frank AML leaves open the possibility that these individuals already had early-stage AML rather than preleukemia.
Two key requirements for any future program to prevent NPM1-mutant AML are (1) the ability to identify NPM1 mutations robustly and reliably and (2) an understanding of their clinical significance in people without overt leukemia. Here, we provide proof-of-principle that NPM1 mutations can be identified in blood DNA of healthy individuals several months prior to the onset of frank AML with 0 false positive calls among the 200 453 WES datasets analyzed. Furthermore, the only 2 individuals with NPM1 mutations in their blood DNA both went on to develop AML, underlying the grave significance of finding these mutations in the blood of healthy individuals. By contrast, “high risk” CH driven by mutations in genes such as U2AF1, SRSF2, TP53, IDH1, and IDH2 does not always progress to myeloid malignancy or only does so after a much longer latency.9,10
Nevertheless, the rarity of NPM1 mutations and the short latency between their identification and AML onset makes the prospect of population screening appear implausible. In fact, recruitment of the 200 453 UKBB participants, aged 38 to 72 years (median 58 years), was undertaken during 2006-2010, and of these, 261 participants developed AML by December 2020 (an incidence of ∼10/100 000 per year, as expected for this age group). More specifically, 15 individuals developed AML in the first year after recruitment (supplemental Figure 2). Although there are no molecular data on AML subtypes among these cases, approximately one-quarter are expected to have been NPM1-mutant, amounting to ∼4 cases in year 1. This aligns well with the fact that we identified only 2 individuals with NPM1 mutations in WES of their blood DNA, both of whom developed AML within 6 months, and proposes that WES is unlikely to detect NPM1 mutations more than 6 months before AML diagnosis. In this context, it is important to consider that the sequencing depth achieved with WES is very shallow. In fact, the depth of coverage of the mutation-bearing final exon of NPM1 in the UKBB was only 18x (95% CI, 6-30x), such that only large clones of NPM1-mutant cells could be detected. Therefore, it is highly probable that deep-targeted NPM1 sequencing would identify individuals with smaller clones that are earlier in disease evolution and may still be in a preleukemic phase. Also, in both cases reported here, AML arose in the context of large DNMT3A-mutant CH clones, proposing that individuals with such clones represent a high-risk group that could be targeted for regular NPM1 mutation screening.
In conclusion, our study demonstrates that NPM1 mutations can be robustly identified in the blood of healthy individuals prior to AML onset but also reveals that shallow sequencing methods such as WES are unlikely to identify NPM1 mutations carriers early enough in disease evolution to facilitate preventive interventions. Future efforts to prevent, delay, or intercept this AML subtype will require much more sensitive approaches able to identify NPM1 mutations in small preleukemic clones. Our findings suggest that such efforts could be focused on individuals with large CH clones, which could in turn be identified through screening of those with subtle abnormalities in their complete blood count results.9 However, large prospective studies are required to enhance/refine screening methodologies and determine the optimal approach to use for the timely identification of preleukemic clones, which would in turn enable clinical studies of targeted interventions to prevent or delay this or other types of myeloid malignancy.11
Acknowledgments: This work was funded by a joint grant from the Leukemia and Lymphoma Society (RTF6006-19), the Rising Tide Foundation (CCR-18-500), and the Wellcome Trust (WT098051). P.M.Q. is supported by the Miguel Servet Program (CP20/00130). G.S.V. is funded by a Cancer Research UK Senior Cancer Fellowship (C22324/A23015), and work in his laboratory is also funded by the European Research Council, Kay Kendall Leukaemia Fund, Blood Cancer UK, and the Wellcome Trust.
Contribution: P.M.Q., M.G., V.I., and C.B. downloaded and analyzed data from the UK Biobank; and G.S.V. conceived and supervised the study.
Conflict-of-interest disclosure: G.S.V. is a consultant for STRM.BIO and Scientific Advisory Board member for Astrazeneca.
Correspondence: George S. Vassiliou, Wellcome-Medical Research Council Cambridge Stem Cell Institute, Department of Haematology, University of Cambridge, Jeffrey Cheah Biomedical Centre, Puddicombe Way, Cambridge CB2 0AW, United Kingdom; e-mail: gsv20@cam.ac.uk.