Key Points
RNA-seq is feasible in the context of a prospective clinical trial for de novo ALL within a clinically sensitive turnaround time.
RNA-seq identified several genetic alterations not detected by conventional methods that confer potential prognostic and therapeutic impact.

Abstract
The molecular hallmark of childhood acute lymphoblastic leukemia (ALL) is characterized by recurrent, prognostic genetic alterations, many of which are cryptic by conventional cytogenetics. RNA sequencing (RNA-seq) is a powerful next-generation sequencing technology that can simultaneously identify cryptic gene rearrangements, sequence mutations and gene expression profiles in a single assay. We examined the feasibility and utility of incorporating RNA-seq into a prospective multicenter phase 3 clinical trial for children with newly diagnosed ALL. The Dana-Farber Cancer Institute ALL Consortium Protocol 16-001 enrolled 173 patients with ALL who consented to optional studies and had samples available for RNA-seq. RNA-seq identified at least 1 alteration in 157 patients (91%). Fusion detection was 100% concordant with results obtained from conventional cytogenetic analyses. An additional 56 gene fusions were identified by RNA-seq, many of which confer prognostic or therapeutic significance. Gene expression profiling enabled further molecular classification into the following B-cell ALL (B-ALL) subgroups: high hyperdiploid (n = 36), ETV6-RUNX1/-like (n = 31), TCF3-PBX1 (n = 7), KMT2A-rearranged (KMT2A-R; n = 5), intrachromosomal amplification of chromosome 21 (iAMP21) (n = 1), hypodiploid (n = 1), Philadelphia chromosome (Ph)-positive/Ph-like (n = 16), DUX4-R (n = 11), PAX5 alterations (PAX5 alt; n = 11), PAX5 P80R (n = 1), ZNF384-R (n = 4), NUTM1-R (n = 1), MEF2D-R (n = 1), and others (n = 10). RNA-seq identified 141 nonsynonymous mutations in 93 patients (54%); the most frequent were RAS-MAPK pathway mutations. Among 79 patients with both low-density array and RNA-seq data for the Philadelphia chromosome-like gene signature prediction, results were concordant in 74 patients (94%). In conclusion, RNA-seq identified several clinically relevant genetic alterations not detected by conventional methods, which supports the integration of this technology into front-line pediatric ALL trials. This trial was registered at www.clinicaltrials.gov as #NCT03020030.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer, constituting ∼25% of annual diagnoses among children under age 15 years.1 Cure rates for childhood ALL have steadily increased over the past 5 decades, and long-term overall survival now exceeds 90% with contemporary chemotherapy regimens.2 This remarkable success culminates from well-designed randomized multi-institutional clinical trials to optimize chemotherapy regimens, enhanced supportive care measures to minimize treatment-related morbidity and mortality, and refinement of risk stratification based on presenting characteristics, leukemia biology, and early disease response to therapy as measured by minimal residual disease (MRD).
Advances in genomics over the last decade, particularly with the advent of next-generation sequencing (NGS) technologies, continue to unravel the genomic landscape of ALL and deepen the understanding of ALL biology. The molecular hallmark of ALL is characterized by recurrent somatic genetic alterations that may carry diagnostic, prognostic and/or therapeutic significance. The constellation of genetic alterations encompasses aneuploidy (eg, hyperdiploidy vs hypodiploidy), chromosomal rearrangements that result in expression of chimeric fusion oncoproteins (eg, BCR-ABL1 and ETV6-RUNX1) or deregulate gene expression, DNA copy number alterations, and sequence mutations.2,3 Current conventional cytogenetic and molecular analyses fail to identify sentinel alterations in ∼25% of childhood ALL.4 In recent years, broad application of NGS technologies, notably whole-transcriptome sequencing (commonly referred to as RNA sequencing [RNA-seq]), has redefined the molecular taxonomy of ALL. RNA-seq enabled discovery of novel genomically defined ALL subtypes characterized by chromosomal rearrangements that are cryptic on karyotyping (eg, DUX4-rearranged [DUX4-R] ALL), myriad gene fusions with numerous partners (eg, MEF2D, ZNF384, or NUTM1-R ALL), and complex and heterogeneous genetic alterations within a single ALL subtype (eg, Philadelphia chromosome-like [Ph-like]) or PAX5 alterations [PAX5alt] ALL).4,5 Thus, RNA-seq offers the ability to simultaneously identify cryptic gene rearrangements, sequence mutations and gene expression profiles in a single assay to accurately determine novel leukemia subtypes along with their drivers, which renders this platform one of the most efficient and clinically relevant among the NGS technologies.
Several nationwide, large-scale precision medicine initiatives, including those from our group, have demonstrated the feasibility of incorporating NGS platforms into expeditious and pragmatic diagnostic, prognostic, and therapeutic algorithms for children with relapsed and/or refractory cancers.6,7 Clinical RNA-seq platforms are becoming increasingly available, but there is little information regarding the use of this technology in the clinical setting.8,9 Herein, we report our experience using RNA-seq to prospectively and in real time molecularly profile a large cohort of consecutive patients with childhood ALL as part of the Dana-Farber Cancer Institute (DFCI) ALL Consortium Protocol 16-001.
Materials and methods
Study design and patient samples
From March 2017 to February 2021, 173 bone marrow or peripheral blood samples were obtained from consecutive children and adolescents age 1 to 21 years with newly diagnosed ALL enrolled on the DFCI ALL Consortium Protocol 16-001 (hereafter DFCI 16-001). The DFCI 16-001 study is a phase 3, multicenter, open-label, randomized clinical trial, testing a novel risk group classification and comparing 2 different pegaspargase dosing regimens after induction (standard fixed dose vs adjusted dose based on nadir serum asparaginase activity levels). The initial risk group was determined at study entry on the basis of age, white blood cell count (WBC), immunophenotype, and central nervous system (CNS) status. Patients who met the following criteria were considered initial low risk (LR): B-cell ALL (B-ALL), younger than age 15 years, WBC < 50 000/μL, and CNS-1 or CNS-2. Patients with any of the following criteria were considered initial very high risk (VHR): IKZF1 deletion, KMT2A-R, low hypodiploidy (≤40 chromosomes), or t(17;19). Final risk group was assigned based on the MRD results. MRD was assessed at 2 time points (TPs) by the clonoSEQ assay (Adaptive Biotechnologies)10 : at day 32 of induction 1A (TP1), and for those with high MRD at TP1, at the end of induction 1B (TP2) (supplemental Figure 1). The study was conducted at 8 pediatric oncology centers in the United States and Canada and was to be closed to the pegaspargase randomization by the end of 2021 with the accrual goal of 480 patients. Diagnostic bone marrow or peripheral blood samples from patients who consented to optional studies on DFCI 16-001 were sent to Centre Hospitalier Universitaire Sainte-Justine (CHUSJ; Montreal, QC, Canada) for sample processing and sequencing. All patient samples were subjected to conventional cytogenetic and molecular analyses to assess for ploidy (karyotype and fluorescence in situ hybridization [FISH]) and the risk-stratifying gene rearrangements by FISH and/or reverse transcriptase polymerase chain reaction (ETV6-RUNX1, TCF3-PBX1, TCF3-HLF, BCR-ABL1, iAMP21, and KMT2A-R) according to institutional standards. A DNA-based NGS assay, the Rapid Heme Panel (RHP), was centrally performed in all patients to detect IKZF1 deletions. The low-density array (LDA) card for Ph-like ALL screening was mandatory for all National Cancer Institute (NCI) patients with high-risk (HR) B-ALL. The protocol was approved by the institutional review board at each participating center. Written informed consent was obtained from parents or legal guardians of all participants.
RNA extraction, library preparation, and sequencing
Total RNA was extracted from the patient’s leukemia cells using mini AllPrep DNA/RNA kits from QIAGEN. TruSeq Stranded Total RNA libraries were prepared by using the Ribo-Zero Gold Kit according to Illumina’s protocol. The resulting libraries (stranded and ribosomal RNA-depleted) were sequenced (paired-end 2 × 75 bp or 2 × 100 bp) on HiSeq 2500, HiSeq 4000, or NovaSeq 6000 systems at the Integrated Center for Clinical Pediatric Genomics at CHUSJ.
Bioinformatic pipelines
RNA-seq analysis was performed as previously described.6 Briefly, alignment to the hg19 (GRCh37) reference genome was performed by using the Spliced Transcripts Alignment to a Reference (STAR) aligner.11 Gene expression was measured with the cufflinks software using the Ensembl version 75 gene coordinates. Point mutations and small insertions/deletions (indels) were identified using the HaplotypeCaller software included in the Genome Analysis Toolkit (GATK) developed at the Broad Institute (Cambridge, MA). Subsequent annotations were added, such as 1000 Genomes Project,12 the Catalogue Of Somatic Mutations In Cancer (COSMIC),13 and predicted effects by Sorting Intolerant From Tolerant (SIFT)14 and Polyphen2 databases.15 Fusion genes, translocations, and chimeric transcripts were identified with FusionCatcher,16 STAR-Fusion,17 and Arriba18 to rank putative reciprocal breakpoints from the STAR output.
Detection of fusions and mutations
Recurrent fusions in ALL, as well as novel fusions implicating 1 known fusion partner were considered for the analysis. Known fusions (available in public databases) or fusions implicating 1 known gene partner in cancers were considered positive when detected by 1 of the 3 tools with at least 2 spanning reads, whereas novel fusions had to be identified by all 3 tools, and they required at least 10 spanning reads. For variant interpretation, the coding regions of a set of 246 leukemia-related genes were analyzed. Filtered-based depth of coverage >30 and allele frequency of <1% in any of the subpopulations of the 1000 Genomes Project database were applied; mutations listed in COSMIC and located within defined genetic hotspots were identified (supplemental Table 2). Novel mutations lying in hotspots and those predicted to be pathogenic by SIFT and Polyphen2 were also reported. Intragenic deletions of IKZF1 were visually detected using the GVIZ R package (version 1.34.0).19
Prediction of ALL subtypes using transcriptional signatures
Hierarchical clustering and experimental t-distributed stochastic neighbor embedding (tSNE) were performed by using the R statistical package and DESeq2 (version 1.30.1).20 We trained the 1-layer neural network (nnet R package, version 7.3-15) on 1134 ALL samples from both in-house (n = 72) and public data sets (n = 1062)21,22 using the top 500 most statistically significant differentially expressed genes among 17 ALL subtypes. The trained model was applied to the sample from each patient in our study, and the subtype with the highest probability by the neural network model was reported as the predicted ALL subtype. In addition, CRLF2 expression was compared with that in the public pediatric ALL TARGET data set and in-house pediatric ALL cohort. CRLF2 expression was considered high when its expression was at the 99th percentile (outlier) with a fragments per kilobase of transcript per million mapped reads (FPKM) value >30. However, the CRLF2 expression was not assessed for all patients but was determined only in patients with CRLF2 fusions or those who had Ph-like gene expression profiles.
Deconvolution of bulk RNA-seq data sets using primary cell types
Results
Patient characteristics and RNA-seq turnaround time
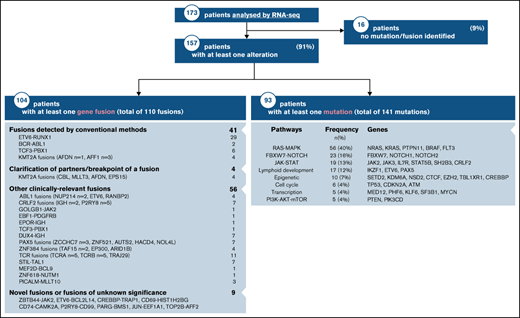
The clinical characteristics of our RNA-seq cohort and those of the overall DFCI 16-001 cohort are shown in Table 1. The median age at diagnosis was 7.2 years (range, 1.0-21.7 years) and the WBC count was 24.6 × 109/L (range, 1.5-874.4 × 109/L) for the RNA-seq cohort. Boys constituted 54.9% of the cohort. There was a predominance of children of the White race (67.6%), whereas patients of Hispanic, Black, or Asian descent represented 13.9%, 5.8%, and 2.9% of the patient cohort, respectively. The majority of patients were CNS-1 (74%) at diagnosis, and only 1 patient presented with testicular involvement. Among 136 patients with B-ALL, 58 (43%) were classified as standard risk (SR), and 78 (57%) as HR according to the NCI Rome criteria. According to the DFCI 16-001 risk stratification, initial risk groups based on presenting clinical features, leukemia immunophenotype, and cytogenetics were low (35.8%), high (43.4%), and very high (20.8%), whereas final risk groups based on initial risk and MRD were low (28.8%), intermediate (28.1%), high (18.1%), and very high (25.0%) for this cohort. As of August 27, 2021, compared with the remainder of the DFCI 16-001 cohort comprising 285 patients, our RNA-seq cohort had a higher median presenting WBC (24.6 vs 9.6 × 109/L; P < .0001), higher proportion of NCI HR patients (65.9% vs 44.0%; P < .001), and higher prevalence of T-cell ALL (T-ALL) patients (21.4% vs 14.0%; P = .053). The comprehensive transcriptomic analysis of 173 unselected patients with de novo ALL (B-ALL, n = 136 [78.6%]; T-ALL, n = 37 [21.4%]) led to the identification of at least 1 genetic alteration in 157 patients (91%) (Figure 1; supplemental Table 1). These alterations included both known and novel fusions genes, nonsynonymous mutations, and intragenic deletions of IKZF1 specifically. The mean time for library preparation, sequencing, bioinformatic analysis, and data interpretation was 19 days (range, 7-48 days). The mean time from sample receipt to final report delivery was 36 days (range, 15-63 days) (Figure 2). The mean time from sample receipt to final report delivery for patients analyzed within the 2017 to 2018 period compared with those analyzed during the 2019 to 2021 period decreased from 38 days to 35 days. The minimal time required from sample receipt to final report completion also shortened to 15 days in 2019 to 2021 compared with 21 days in 2017 to 2018.
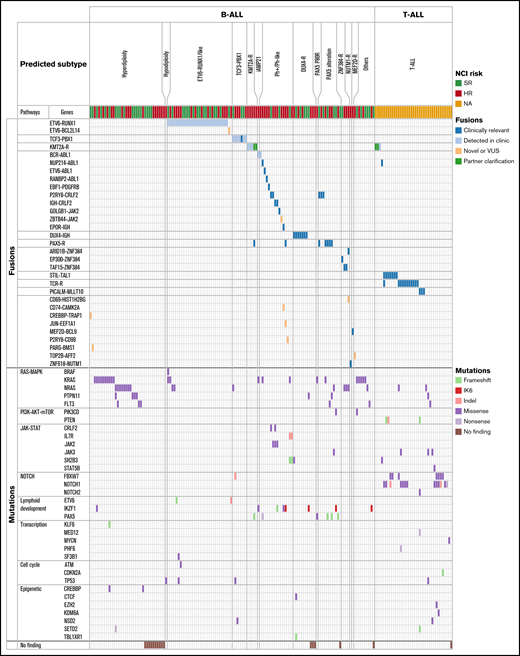
Comprehensive heatmap of molecular profiling findings by RNA-seq of 173 patients with ALL enrolled on DFCI 16-001. RNA-seq data from 173 patients with ALL are summarized in this heatmap. Patients were classified on the basis of their respective clustering analysis subtype, gene fusions, and somatic mutations. Specific gene fusions and mutational categories are indicated by colored squares and classified by associated gene signaling pathways. Mutation types included single nucleotide variants, indels, and intragenic IKZF1 deletion (IK6). iAMP21, intrachromosomal amplification of chromosome 21; NA, not applicable; TCR-R, T cell receptor rearrangement; VUS, variant of unknown significance.
Comprehensive heatmap of molecular profiling findings by RNA-seq of 173 patients with ALL enrolled on DFCI 16-001. RNA-seq data from 173 patients with ALL are summarized in this heatmap. Patients were classified on the basis of their respective clustering analysis subtype, gene fusions, and somatic mutations. Specific gene fusions and mutational categories are indicated by colored squares and classified by associated gene signaling pathways. Mutation types included single nucleotide variants, indels, and intragenic IKZF1 deletion (IK6). iAMP21, intrachromosomal amplification of chromosome 21; NA, not applicable; TCR-R, T cell receptor rearrangement; VUS, variant of unknown significance.
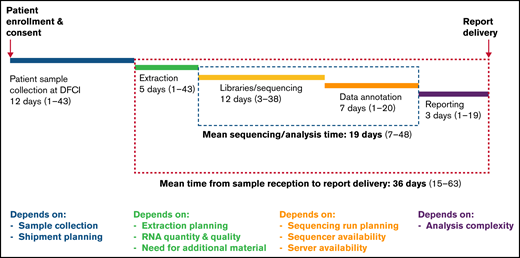
Timeline of the clinical implementation of RNA-seq from sample receipt to report delivery and factors contributing to timeline variation at each step.
Timeline of the clinical implementation of RNA-seq from sample receipt to report delivery and factors contributing to timeline variation at each step.
Detection of fusion genes
The median coverage depth by RNA-seq was 179 million read-pairs (range, 63-406 read-pairs). Using this data set, we detected at least 1 expressed gene fusion in 104 (60%) of the 173 patients tested for a total of 110 fusions. Only 37% (41 of 110) of gene fusions identified by RNA-seq would have been identified by conventional cytogenetic modalities. Fusion detection by RNA-seq was 100% concordant with the results obtained from conventional cytogenetic analyses for all 41 fusion genes detected in the clinic. In 4 additional patients, RNA-seq allowed the identification of partner genes or breakpoints for a gene rearrangement identified by FISH in the clinic (eg, KMT2A-R). In addition to recurrent fusion genes that are routinely tested for in the clinic (ETV6-RUNX1, BCR-ABL1, TCF3, or KMT2A-r by FISH), we uncovered 56 clinically relevant gene fusions by RNA-seq. Some of these gene fusions have recently been associated with prognostic or therapeutic significance. For instance, DUX4-r has been associated with a favorable outcome,21 and ZNF384-r and PAX5-r have been associated with an intermediate prognosis.25,26 In addition, several kinase-activating fusions involving ABL1, PDGFRB, CRLF2, or JAK2 could be targeted by therapy with specific tyrosine kinase inhibitors.27 RNA-seq also led to the identification of novel oncogenic fusions (eg, the ZBTB44-JAK2 [exon 2-exon 19]) in-frame fusion and its reciprocal product (identified in a patient with Ph-like ALL) that contained the N‐terminal protein‐interacting BTB domain of ZBTB44 and the kinase domain of JAK2 (Figure 3).
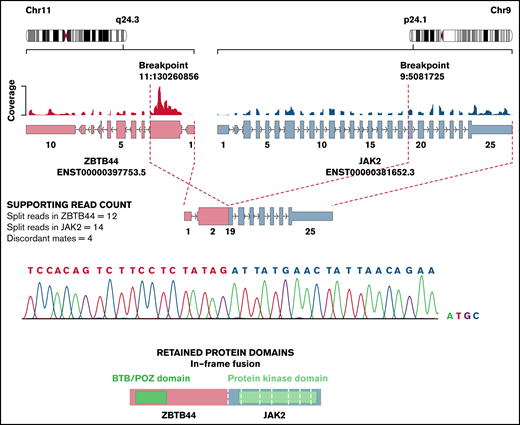
Novel ZBTB44-JAK2 fusion detected in a patient with Ph-like ALL. Exon 2 of ZBTB44 fused in-frame to exon 19 of JAK2, conserving an intact kinase domain. The fusion was validated by reverse transcriptase polymerase chain reaction and Sanger sequencing. The figure was adapted from Arriba output.
Novel ZBTB44-JAK2 fusion detected in a patient with Ph-like ALL. Exon 2 of ZBTB44 fused in-frame to exon 19 of JAK2, conserving an intact kinase domain. The fusion was validated by reverse transcriptase polymerase chain reaction and Sanger sequencing. The figure was adapted from Arriba output.
IKZF1 intragenic deletion
An increasing body of clinical evidence demonstrates that the presence of IKZF1 deletion is associated with adverse outcome in children and adult ALL.28-32 Among 25 patients with IKZF1 deletions identified by the RHP, RNA-seq identified only the intragenic deletion of exons 4 to 7 of IKZF1 resulting in the dominant-negative IK6 isoform in 4 patients (Ph-like, n = 1; DUX4-R, n = 1; PAX5alt, n = 1; B-Others, n = 1) (Figure 4). Combining gene expression signature from RNA-seq and IKZF1 deletion status from the RHP, IKZF1 deletion was found in patients with Ph-like ALL (n = 10), PAX5alt (n = 3), B-Others (n = 2), ZNF384-R (n = 1), BCR-ABL1 (n = 1), TCF3-PBX1 (n = 1), and 28% of favorable subtypes (high hyperdiploid [HHD], n = 5; DUX4-R: n = 2).
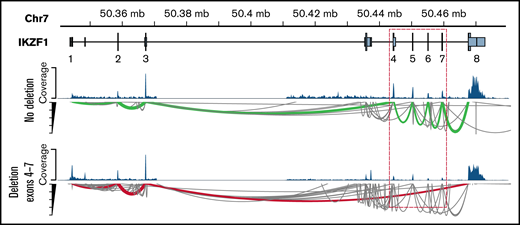
Detection of intragenic IKZF1 deletions by RNA-seq. Intragenic deletions of exons 4 to 7 of IKZF1 visualized using the GVIZ R package. Green line corresponds to normal IKZF1 transcript (IK1), and the red line corresponds to IK6 transcript missing exons 4 to 7 (example with patient #22).
Detection of intragenic IKZF1 deletions by RNA-seq. Intragenic deletions of exons 4 to 7 of IKZF1 visualized using the GVIZ R package. Green line corresponds to normal IKZF1 transcript (IK1), and the red line corresponds to IK6 transcript missing exons 4 to 7 (example with patient #22).
Detection of mutations
Using RNA-seq data, we identified a total of 141 sequence mutations in a list of 246 leukemia-related genes. At least 1 pathogenic mutation was found in 93 patients (54%) (Figure 1). Among these, 108 were distinct mutations, including 20 that were recurrent in our cohort (supplemental Table 3). Seventy-nine patients (85%) harbored a mutation that carries a potential prognostic and/or therapeutic significance. We identified recurrent mutations in genes or related signaling pathways including RAS-MAPK, JAK-STAT, FBXW7/NOTCH, and epigenetics that could provide a rationale for targeted therapies. RAS-MAPK pathway mutations were most prevalent within the HHD subtype. The alterations identified in this study were not mutually exclusive, as illustrated by patients who carried several mutations in their cancer. The most frequently mutated signaling pathway was the RAS-MAPK pathway with 56 mutations, which represented 40% of all detected mutations. Most mutations were listed in COSMIC.13 We detected 21 novel potentially oncogenic mutations that were located within known functional hotspots and/or that predicted damage that would require further functional characterization.
Molecular classification of known and novel ALL subtypes using gene expression signature
We developed gene expression profiling tools using hierarchical clustering, tSNE analysis, and a trained 1-layer neural network (Figure 5) to predict different ALL subtypes using RNA-seq data. We applied the molecular classifier to each of the 173 patients with ALL. This allowed the classification of patients into 1 of the following 17 subtypes: HHD (n = 36), ETV6-RUNX1 (n = 29), TCF3-PBX1 (n = 7), KMT2A-R (n = 5), BCR-ABL1 (n = 1), iAMP21 (n = 1), hypodiploid (n = 1), Ph-like (n = 15), ETV6-RUNX1-like (n = 2), DUX4-R (n = 11), PAX5alt (n = 11), PAX5 P80R (n = 1), ZNF384-R (n = 4), NUTM1-R (n = 1), MEF2D-R (n = 1), B-Others (n = 10), and T-ALL (n = 37) (Figure 1). Using transcriptional signatures from RNA-seq data, it was possible to reclassify 42 of 52 B-others into novel molecular subtypes based on subtype-defining alterations, leaving 10 patients (7%) as truly B-Others (supplemental Figure 2).
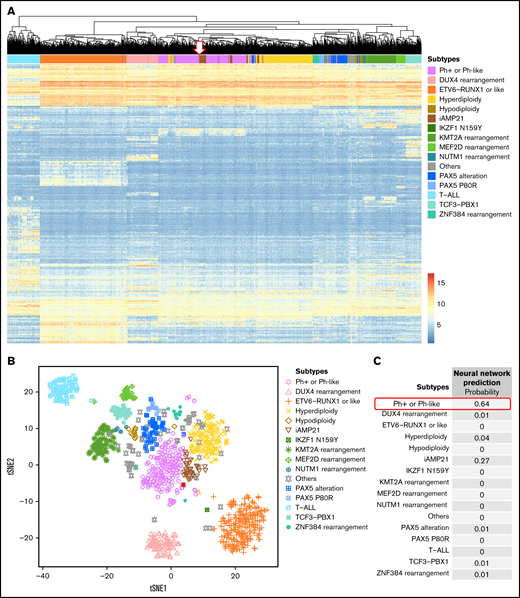
Molecular subtype clustering of patients with ALL based on gene expression signatures. (A) Hierarchical clustering and (B) experimental t-distributed stochastic neighbor embedding (tSNE) performed using the top 500 variable genes from 1134 ALL samples from both in-house and public RNA-seq data sets. (C) The neural network probability score for each of the subtypes listed for panels A-C. Results for patient #124 are shown by the arrow in A and the red dot in B.
Molecular subtype clustering of patients with ALL based on gene expression signatures. (A) Hierarchical clustering and (B) experimental t-distributed stochastic neighbor embedding (tSNE) performed using the top 500 variable genes from 1134 ALL samples from both in-house and public RNA-seq data sets. (C) The neural network probability score for each of the subtypes listed for panels A-C. Results for patient #124 are shown by the arrow in A and the red dot in B.
Ph-like gene classifier by LDA card vs RNA-seq
The LDA card is a screening assay for patients with the Ph-like kinase-activated gene signature33,34 used in the Children’s Oncology Group (COG) and other North American consortia and is mandatory for all patients with NCI HR B-ALL enrolled on the DFCI 16-001 protocol. RNA-seq analysts were blinded to the LDA results, and results from both assays were compared for concordance. Among 79 patients with both LDA and RNA-seq data for the Ph-like gene signature prediction, the results were concordant in 74 (94%) of 79 patients. A total of 5 patients had discordant results with a positive score by LDA and a negative score by RNA-seq for the Ph-like gene signature for which 3 of 5 patients could represent LDA’s false-positive patients. The first patient (#57) with a discordant result was identified as hypodiploid subtype with a TP53 mutation, whereas the other 2 discordant patients (#130 and #156) were HHD patients without kinase-activating alterations by RNA-seq. Low-hypodiploid and HHD ALL subtypes had distinct expression signatures and are typically mutually exclusive with Ph-like ALL.4 RNA-seq failed to identify 2 patients (#96 and #167) with CRLF2 overexpression and P2RY8-CRLF2 fusions. Furthermore, we identified 3 Ph-like patients (5%) among 59 patients with NCI SR B-ALL, which supports the clinical relevance of an unbiased screening approach. Of importance, RNA-seq detected targetable kinase-activating alterations in 11 (73%) of 15 patients with Ph-like ALL.
Molecular classification of T-ALL
Among 37 patients with T-ALL, 25 gene fusions and 47 sequence mutations were identified in 36. Twenty-four patients (67%) harbored gene fusions that could be subdivided into the following categories: T-cell receptors and transcription factor oncogenes (n = 17), KMT2A-r (n = 3), PICALM-MLLT10 (n = 3), and ABL1-r (n = 1). Five of the 8 molecular subgroups previously reported in T-ALL35 could be classified by the presence of fusions involving the following genes: TAL1 (n = 7), TLX1 (n = 2), LYL1 (n = 1), TLX3 (n = 1), and HOXA (n = 1). Mutations were identified in 7 of the 10 most frequently dysregulated functional pathways in T-ALL35 : NOTCH (n = 16), JAK-STAT (n = 5), Ras (n = 4), epigenetic (n = 4), PI3K-AKT (n = 3), transcription (n = 3), and cell cycle or tumor suppression (n = 2).
Determination of blood cell types
We used DeconRNASeq to estimate the proportions of both B- and T-cell types by deconvoluting blood cell types using patients’ bulk expression data (Figure 6). Immunophenotype was concordant between clinical conventional assessment and deconvoluted cell analysis in 142 (82%) of 173 patients. Among the 31 discordant cases, 16 (52%) were T-ALL, and 15 (48%) were B-ALL. Among 170 samples with available blast percentages provided by the clinical laboratory, 66 samples (39%) had more than 25% discordance between the deconvoluted cell type proportions by DeconRNASeq and the clinical blast count. Although all samples had a clinical blast count >25%, 28 samples (16%) had <25% blast count estimated by DeconRNASeq, of which an alteration was detected in 26 (93%) of 28 samples. RNA-seq did not detect any alteration in 13 samples, of which 2 (15%) had a blast count <25% by DeconRNASeq.
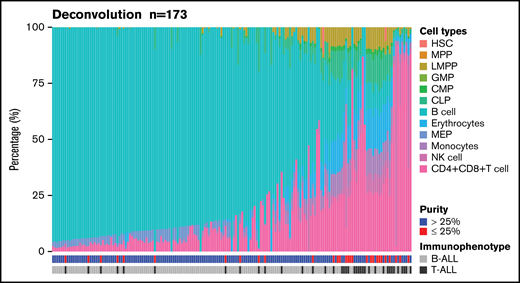
Estimation of blood cell populations using a deconvolution tool. Proportion of primary hematopoietic cell types in 173 patients with ALL using 13 primary cell-type expression profiles. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GMP, granulocyte-monocyte progenitor; HSC, hematopoietic stem cell; LMPP, lymphoid-primed multipotential progenitor; MEP, megakaryocyte-erythrocyte progenitor; MPP, multipotent progenitor; NK cell, natural killer cell.
Estimation of blood cell populations using a deconvolution tool. Proportion of primary hematopoietic cell types in 173 patients with ALL using 13 primary cell-type expression profiles. CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GMP, granulocyte-monocyte progenitor; HSC, hematopoietic stem cell; LMPP, lymphoid-primed multipotential progenitor; MEP, megakaryocyte-erythrocyte progenitor; MPP, multipotent progenitor; NK cell, natural killer cell.
Discussion
Our study demonstrated the feasibility and clinical utility of RNA-seq as an integrative diagnostic assay in the molecular classification and prognostic stratification of an unbiased cohort of de novo childhood ALL. We analyzed whole-transcriptome data from 173 consecutive childhood ALL patients and identified at least 1 genetic alteration in 91% of them that may carry diagnostic, prognostic, and/or therapeutic significance. There was a 100% concordance of gene fusion detection between RNA-seq and conventional cytogenetics. This high concordance level might be explained by the deep coverage and the accurate fusion calls enabled by the combination of 3 predicting algorithms (FusionCatcher, STAR-Fusion, Arriba). Although RNA-seq was not used for DFCI-defined risk-stratifying criteria, RNA-seq enabled the identification of 56 clinically relevant gene fusions not detected by conventional cytogenetics, and it accurately identified novel molecular subtypes with prognostic and/or therapeutic significance, especially Ph-like ALL, thus reducing the proportion of the B-Others subgroup by conventional cytogenetics from 39% to 7%. In addition to fusion transcripts and gene expression profiles, RNA-seq also identified clinically relevant mutations in more than half the patients in our cohort. We further showed that centralized RNA-seq was feasible within a prospective multi-institutional trial; results were returned within a mean of 36 days from sample receipt. It is worth mentioning that samples were batched at different steps from sample shipping to report delivery, which accounts for the wide range in the timeline that could be significantly shortened by direct sample processing and sequencing. Indeed, the minimal time required to complete the final report from sample receipt was 15 days for samples analyzed within the 2019 to 2021 study period.
Because subtype-defining chromosomal alterations in ALL are heterogeneous and/or cryptic with conventional karyotype analysis, RNA-seq offers an integrated platform to provide leukemic gene expression signatures, known and novel chromosomal rearrangements, and sequence mutations, each of which greatly contributes to ALL profiling and refining risk stratification. For instance, several groups, including ours, are currently using IKZF1 deletions for risk stratification and treatment intensification.36,37 Nevertheless, IKZF1 alterations are enriched in Ph+ ALL and Ph-like ALL which are associated with an unfavorable outcome. But they are also prevalent in DUX4-R and ETV6-RUNX1-like ALL which confer a favorable prognosis.21 Therefore, RNA-seq provides a global genomic landscape for each patient rather than a single alteration that may not allow for accurate molecular classification and risk stratification. RNA-seq allows for ongoing discovery of novel gene fusions, especially among the Ph-like ALL subtype, which is characterized by a heterogeneous spectrum of kinase fusions.27 In our cohort, we identified the ZBTB44-JAK2 fusion, which has not been reported thus far. ZBTB44 is a member of the zinc finger and BTB/POZ domain–containing protein (ZBTB) family that is involved in both normal and malignant hematopoiesis.38 A similar ZBTB44 fusion with the kinase FLT3 was previously found in a 64-year-old man with unclassified myeloproliferative neoplasms and hypereosinophilia.39 Other members of the ZBTB family were reported as 5′ partners of JAK2 fusions in ALL (eg, ZBTB20-JAK2 and ZBTB46-JAK2).40,41 To date, more than 20 different partner genes have been identified with JAK2 fusions in Ph-like ALL; all JAK2 fusions conserved an intact kinase domain and conferred constitutive JAK2 kinase activation.41
Another advantage of RNA-seq that we demonstrated is the ability to identify clonal mutations at diagnosis that may carry prognostic and therapeutic impact. For example, RAS-MAPK pathway mutations and mutations in genes involved in drug metabolism, which are often enriched in relapsed ALL, were identified at diagnosis in our cohort. We and others have shown that patients with HHD ALL with clonal RAS or CREBBP mutations at diagnosis had a worse outcome compared with their counterparts without clonal RAS or CREBBP mutations.42 Therefore, early detection of these variants at diagnosis could further refine risk stratification in HHD which is usually associated with favorable prognosis but still contributes to a significant proportion of relapses in the current treatment era. Regarding T-ALL, those harboring PI3K pathway mutations had an inferior prognosis as previously reported by our group43 and others.44,45 Furthermore, RNA-seq identified several dysregulated signaling pathways in T-ALL that may represent novel therapeutic targets for a patient population in whom treatment of primary refractory and recurrent disease remains suboptimal. Several signal transduction inhibitors targeting JAK-STAT,46 Ras-MAPK,47 or PI3K-AKT-mTOR48,49 pathways have been investigated in preclinical models and early-phase trials. Despite the initial failure of gamma-secretase inhibitors (GSIs) in treating NOTCH1-activated tumors because of insufficient efficacy and excessive gastrointestinal toxicity, ongoing efforts are underway to target NOTCH because it is the most frequently mutated pathway in T-ALL. For example, the combination of corticosteroids and GSIs for mitigating gastrointestinal toxicity and enhancing antitumor activity are being evaluated in clinical trials in addition to newer, more selective GSIs such as PSEN1 inhibitors50 or NOTCH1 monoclonal antibodies.51 Incorporating RNA-seq could thus facilitate molecular-based risk stratification and precision medicine opportunities for specific T-ALL subsets.
Although the comprehensive ALL profiling by RNA-seq enables the identification of novel molecular subtypes, Ph-like ALL is currently the most important clinically relevant subtype that cannot be easily identified by conventional cytogenetic techniques. The primary strategy currently used by the COG for Ph-like ALL screening is the LDA card.33 We have demonstrated the high concordance of the LDA assay and RNA-seq to accurately identify patients with a Ph-like gene expression profile. However, RNA-seq offers a greater advantage than the LDA card itself because RNA-seq simultaneously detected the underlying kinase-activation alteration that may be therapeutically targetable in 11 (73%) of 15 of patients with Ph-like ALL. Furthermore, 3 NCI patients with Ph-like ALL in our cohort had SR B-ALL, which would further support Ph-like ALL screening in patients with either NCI SR or HR ALL.
In this study, we pilot the use of a deconvolution tool (DeconRNASeq) into our RNA-seq pipelines to determine different cell-type populations, which may serve multiple functions. First, the tool evaluates the true blast content of the sequenced sample, because the bone marrow sample received for molecular sequencing often had a different blast percentage than what had been determined in the clinic as a result of consecutive bone marrow aspirations (eg, first aspirate vs subsequent pull). Low blast content may fail to detect sequence mutations by RNA-seq. Among 66 samples with either a 25% discordance rate in blast percentage or having <25% of blasts evaluated by DeconRNASeq, 61 samples (92%) still had an alteration detected by RNA-seq, but only 27 (41%) were found to have a sequence mutation. In the future, this tool could be optimized to further determine which patients have mixed-phenotype acute leukemia, an entity in which lineage plasticity may render the immunophenotypic interpretation difficult, or to assess the immune repertoire of patients with ALL at specific time points during treatment.
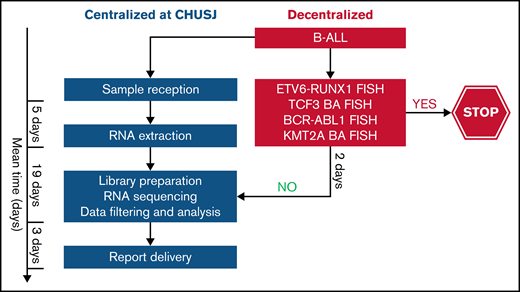
As noted in other reports, RNA-seq is not optimal for assessing copy number alterations.52 The assessment of IKZF1 deletions by RNA-seq in our study is limited to the dominant-negative IKZF1 isoform. The full spectrum of IKZF1 deletions may be complemented by other diagnostic assays such as microarray comparative genomic hybridization37 or the RHP, which is currently used in the DFCI 16-001 protocol.53 In addition, RNA-seq cannot detect the intragenic amplification of PAX5, which is another alteration found within the PAX5alt subtype. This may explain the absence of PAX5 alterations in 4 of the 11 patients with PAX5alt predicted by RNA-seq. Other limitations of RNA-seq include longer turnaround time and costs that are mutually related; however, these limitations can be overcome over time as the clinical algorithms become standardized, and NGS can replace some of the more labor-intensive cytogenetic tests. Alternatively, we propose a tiered algorithm in which RNA-seq could be incorporated after a restricted FISH panel of relatively frequent and prognostically relevant fusions (ETV6-RUNX1, BCR-ABL1, TCF3, or KMT2A-r) to rapidly exclude recurrent alterations associated with a favorable prognosis and/or those that are mutually exclusive with subtypes that are primarily detected by RNA-seq or other molecular techniques (Figure 7). By using this algorithm, we would exclude a third of our patient cohort that required further genomic characterization and enrich our cohort to identify those with subtype-defining alterations by RNA-seq.
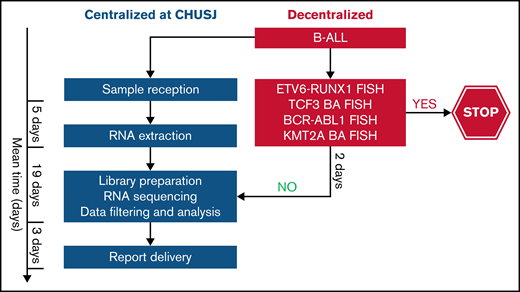
Proposed tiered algorithm for a time- and cost-effective clinical implementation of RNA-seq. Proposed algorithm incorporating FISH and RNA-seq with an estimated turnaround time of 4 weeks from the time of sample receipt to final report delivery based on our experience. BA FISH, break-apart fluorescence in situ hybridization.
Proposed tiered algorithm for a time- and cost-effective clinical implementation of RNA-seq. Proposed algorithm incorporating FISH and RNA-seq with an estimated turnaround time of 4 weeks from the time of sample receipt to final report delivery based on our experience. BA FISH, break-apart fluorescence in situ hybridization.
In conclusion, RNA-seq is feasible in the context of a multicenter prospective clinical trial within a clinically sensitive turnaround time. We further identified several clinically relevant genetic alterations that were not detected by conventional methods, which supports integrating this technology into front-line trials for childhood ALL. The lessons learned from this experience provided opportunities to optimize our bioinformatic pipelines, standardize variant interpretation, and lay the foundation for implementing RNA-seq in the clinic. With longer follow-up, future analysis will focus on describing the outcomes of our DFCI 16-001 patient cohort when stratified by RNA-seq–based molecular subtypes to further determine the clinical value of RNA-seq.
Acknowledgments
The authors are indebted to the patients and their parents for participating in this study. Patient tissue samples were provided by the Centre Hospitalier Universitaire (CHU) Sainte-Justine Pediatric Cancer biobank. NGS was performed at the Integrated Clinical Genomics Centre in Pediatrics, CHU Sainte-Justine. Computations were made on the supercomputer managed by Calcul Québec and Compute Canada.
This study was supported by the Fondation Charles-Bruneau, Cole Foundation, and the Leukemia & Lymphoma Society of Canada.
D.S. holds the François-Karl-Viau Research Chair in Pediatric Oncogenomics.
Authorship
Contribution: T.H.T., D.S., and L.B.S. designed and oversaw the study; P.S.-O. and M.C. developed the bioinformatic pipelines; T.S. processed all of the samples; S.L., C.M., C.J.-C., and A.R. analyzed the genomic data; K.M.S., V.K., and K.E.S. collected and analyzed the clinical data; T.H.T., J.-M.L., H.B., C.L., V.-P.L., S.C., P.D.C., L.M.G., J.M.K., K.M.K., R.S., B.M., J.J.G.W., and L.B.S. recruited patients; T.H.T., S.L., C.M., A.R.B., M.C., P.S.-O., and D.S. prepared the manuscript; and all authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thai Hoa Tran, Centre Hospitalier Universitaire Sainte-Justine, 3175 Chemin de la Côte-Sainte-Catherine, Montréal, QC H3T 1C5, Canada; e-mail: thai.hoa.tran@umontreal.ca; and Daniel Sinnett, Centre Hospitalier Universitaire Sainte-Justine, 3175 Chemin de la Côte-Sainte-Catherine, Montréal, QC H3T 1C5, Canada; e-mail: daniel.sinnett@umontreal.ca.