

Key Points
Low-dose TBI added to allo-HCT conditioning does not further increase the risk of SM.
A greater number of pretransplant chemotherapy cycles are associated with an increased risk of SM after allo-HCT.

Abstract
Subsequent malignancies (SMs) present a significant burden of morbidity and are a common cause of late mortality in survivors of allogeneic hematopoietic cell transplant (allo-HCT). Previous studies have described total body irradiation (TBI) as a risk factor for the development of SMs in allo-HCT survivors. However, most studies of the association between TBI and SM have examined high-dose TBI regimens (typically 600 cGy), and thus little is known about the association between low-dose TBI regimens and risk of SMs. Our goal, therefore, was to compare the cumulative incidence of SMs in patients of Alberta, Canada, who received busulfan/fludarabine alone vs busulfan/fludarabine plus 400 cGy TBI. Of the 674 included patients, 49 developed a total of 56 malignancies at a median of 5.9 years’ posttransplant. The cumulative incidence of SMs at 15 years’ post-HCT in the entire cohort was 11.5% (95% confidence interval [CI], 8.5-15.6): 13.4% (95% CI, 9.1-19.3) in the no-TBI group and 10.8% (95% CI, 6.6-17.4) in the TBI group. In the multivariable model, TBI was not associated with SMs, whereas there was an association with number of pre-HCT cycles of chemotherapy. The standardized incidence ratio for the entire cohort, compared with the age-, sex-, and calendar year–matched general population, was 1.75. allo-HCT conditioning that includes low-dose TBI does not seem to increase risk of SMs compared with chemotherapy-alone conditioning.
Introduction
Subsequent malignancies (SMs) are a well-described late toxicity of allogeneic hematopoietic cell transplant (allo-HCT), occurring at more than twice the expected rate in survivors compared with the general population.1,2 SMs present a significant burden of morbidity and are a common cause of late mortality in survivors of allo-HCT.3,4 The cumulative incidence of SMs after allo-HCT varies depending on population studied and study methodology but is reportedly as high as 15% at 15 years’ post-HCT, with an ever-increasing incidence posttransplant and no observable plateau.2,5 Previous studies have described total body irradiation (TBI) as a risk factor for the development of SMs in allo-HCT survivors: a dose-dependent relationship between TBI and SM risk has been described, with risk of SM increasing with cumulative dose and decreasing with the use of fractionation.1,6-8 However, most studies of the association between TBI and SM have examined high-dose TBI regimens (typically 600 cGy), and thus little is known about the association between low-dose TBI regimens (typically <600 cGy) and risk of SMs.
In recent years, lower dose TBI has been incorporated into myeloablative and non-myeloablative conditioning regimens to reduce toxicity while retaining the immunosuppressive and antileukemic properties of TBI.9-12 Determining the potential late toxicities associated with low-dose TBI is critical, both for counseling patients pretransplant and for delivering effective survivorship care. In Alberta, Canada, starting in ∼2003, 400 cGy of TBI was added to our standard myeloablative busulfan/fludarabine conditioning regimen with the intention of reducing the risk of posttransplant relapse.13 It is imperative to understand whether low-dose TBI places patients at an increased risk of SM. Thus, our goal was to compare the cumulative incidence of SM in patients of Alberta who received busulfan/fludarabine alone vs busulfan/fludarabine plus 400 cGy TBI.
Methods
Patients
This study included sequential patients who received allo-HCT in Alberta between January 1, 1999, and December 31, 2014, who received standard conditioning with fludarabine/busulfan with or without 400 cGy TBI and who survived until at least 1 year posttransplant. Those who received a conditioning regimen other than fludarabine/busulfan and those who died before 1 year posttransplant were excluded. Patients were followed up post-HCT at either the Cross Cancer Institute in Edmonton or the Tom Baker Cancer Centre in Calgary. The study was approved by the Health Research Ethics Board of the Alberta Cancer Committee and was conducted in accordance with the Declaration of Helsinki.
Conditioning regimens and transplant
Conditioning and transplant details have been described in detail elsewhere.10,13 Briefly, conditioning consisted of fludarabine (50 mg/m2 per day IV on days −6 to −2), busulfan (∼3.2 mg/kg per day IV on days −5 to −2; since 2010, pharmacokinetically adjusted to target daily exposure of 3750 μmol/L × min), and antithymocyte globulin (Thymoglobulin, 4.5 mg/kg IV; 0.5 mg/kg on day −2, 2 mg/kg on day −1, and 2 mg/kg on day 0) with or without TBI (400 cGy in 2 fractions on day −1). Additional graft-versus-host disease (GVHD) prophylaxis consisted of cyclosporine and short-course methotrexate. Before 2003, TBI was added to conditioning for those with acute lymphoblastic leukemia or for those with any acute leukemia with extramedullary disease. In 2003, TBI was added to conditioning for all patients with acute leukemia. Finally, between 2009 and 2012, TBI was added to conditioning for all patients with hematologic malignancies.
Subsequent malignancies
All SMs were included except: (1) relapse of the malignancy for which allo-HCT was undertaken; (2) relapse or progression of a malignancy that existed before allo-HCT; (3) cervical intraepithelial neoplasia; (4) non-melanoma skin cancers; and (5) posttransplant lymphoproliferative disorder. SMs were identified for each included patient through a search of the Alberta Cancer Registry (ACR).14 The ACR is a central repository for the province; every diagnosis of malignancy in Alberta is reported to the registry. After data pull from the ACR, each reported malignancy was reviewed by the authors to ensure that no exclusion criteria, as outlined earlier, were present. For rare patients who moved out of the province posttransplant but continued follow up in Alberta, medical records were reviewed to identify SMs.
Statistical analysis
Comparisons within the cohort.
The cumulative incidence of SM in the entire cohort, in those who received TBI, and in those who did not receive TBI was estimated taking into account competing risks. Competing risks included death without SM and second allo-HCT. A multivariable Cox regression model using age as the time scale (to control for increasing risk of malignancy with increasing age)15 was used to assess the association of receipt of low-dose TBI and SM. Subjects were entered into the model at 1 year posttransplant and were censored at the earliest of last follow-up, second HCT, or death. Covariates were selected a priori and included grades II to IV acute GVHD (aGVHD), moderate to severe chronic GVHD (cGVHD), age at transplant (in years), sex, graft type (peripheral blood stem cell vs other), underlying diagnosis (lymphoma/chronic lymphocytic leukemia [CLL] vs others), number of chemotherapy cycles before HCT, and history of localized radiotherapy before HCT. Chemotherapy and radiotherapy used for the treatment of any malignancy that existed before HCT was included; for example, if HCT was undertaken for therapy-related leukemia after treatment of a solid tumor, chemotherapy and radiotherapy for the solid tumor and for the leukemia were included. Targeted therapies such as monoclonal antibodies and tyrosine kinase inhibitors, when used as single agents, were not counted as pre-HCT chemotherapy. For single-agent oral cytotoxic therapies (eg, chlorambucil) without specified cycle lengths, 2 months of therapy was counted as one cycle. To rule out the potential impact of the pharmacokinetic adjustment of busulfan that began in 2010, the regression model was repeated with only those undergoing transplant before 2010 included in the model.
Comparisons vs the general population.
Standardized incidence ratios (SIRs) were calculated as follows: expected numbers of cancers for the entire cohort and various subpopulations of the cohort were calculated by applying age-, sex-, and calendar year–matched Alberta-specific cancer incidence rates (obtained from Statistics Canada)16 to total person years at risk in the cohort or subpopulation of the cohort. The SIR is the ratio of observed to expected malignancies. Confidence intervals (CIs) and significance testing for SIRs were calculated assuming a Poisson distribution. Excess absolute risk (EAR) was calculated as follows: the observed number of cancers minus the expected number of cancers, divided by person years at risk, and expressed as excess cancers per 1000 person years at risk.
Results
A total of 1011 patients received an allo-HCT between January 1, 1999, and December 31, 2014. Of these, 337 were excluded from study, including 56 who received a nonstandard chemotherapy protocol and 281 who died before reaching 1 year posttransplant. Of the remaining 674 patients, 422 received TBI, and 252 did not. The characteristics of both cohorts are shown in Table 1. This was predominantly an adult cohort, with a median age of 47 years (interquartile range [IQR], 36-56 years). Median follow-up among all survivors was 9.4 years (IQR, 6.7-13.0 years): 11.7 years (IQR, 9.0-15.1 years) in the no-TBI group and 8.2 years (IQR, 6.0-11.4 years) in the TBI group. The vast majority of patients in each group received allo-HCT for hematologic malignancies. Of these, most were for lymphomas (38%) in the no-TBI group and for leukemias (79%) in the TBI group. The majority of allo-HCTs were from matched donors (90% in the no-TBI group and 84% in the TBI group) and used peripheral blood stem cells (80% in the no-TBI group and 93% in the TBI group).
Subsequent malignancies
Overall, 49 patients developed a total of 56 malignancies at a median of 5.9 years’ posttransplant (IQR, 3.4-9.7 years). A variety of sites and histology of malignancy sites were observed (Table 2). The most common sites of malignancy were gastrointestinal (n = 13) and genitourinary (n = 13). Adenocarcinomas accounted for 24 malignancies, and squamous cell carcinomas accounted for 16.
Cumulative incidence
The cumulative incidence of SM at 15 years’ post-HCT in the entire cohort was 11.5% (95% CI, 8.5-15.6) (Figure 1): 13.4% (95% CI, 9.1-19.3) in the no-TBI group and 10.8% (95% CI, 6.6-17.4) in the TBI group (Figure 2).
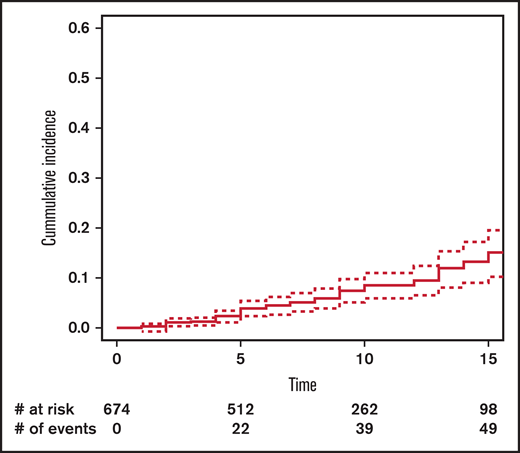
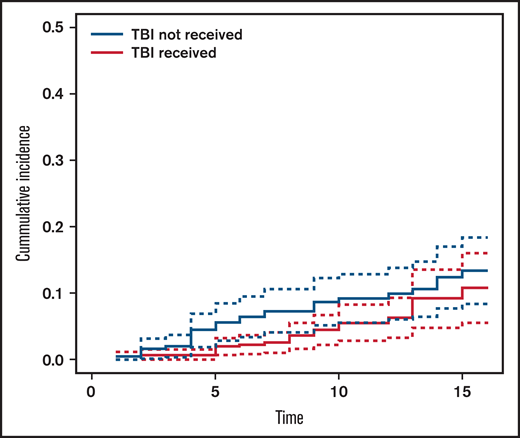
Cumulative incidence of subsequent malignancy by exposure to total body irradiation (TBI).
Cumulative incidence of subsequent malignancy by exposure to total body irradiation (TBI).
Within-cohort comparisons
Results of the multivariable Cox regression model are shown in Table 3. Receipt of TBI was not associated with SM (hazard ratio [HR], 0.81; 95% CI, 0.51-3.73; P = .52). The only highly significant association with SM was pre-HCT cycles of chemotherapy (HR of 1.07 for each cycle of chemotherapy before HCT; 95% CI, 1.01-1.12; P = .01). The occurrence of moderate to severe cGVHD (HR, 1.65; 95% CI, 0.88-3.08; P = .12) seemed to be associated with SM, although not statistically significant. The remaining covariates (grades II-IV aGVHD, recipient sex, age at HCT, underlying disease, pre-HCT localized radiotherapy, and graft type) were not associated with SM.
Of the entire cohort, 431 patients received allo-HCT before pharmacokinetic adjustment of busulfan (ie, before 2010): 215 received TBI and 216 did not receive TBI. When the regression model was restricted to these patients, there remained no association between receipt of TBI and SM (HR, 0.82; 95% CI, 0.36-1.89; P = .65). Pre-HCT cycles of chemotherapy remained significantly associated with SM (HR, 1.10; 95% CI, 1.04-1.18; P = .002). As in the analysis of the full cohort, the remaining covariates were not associated with SM.
Comparisons vs the general population
SIRs and EARs are detailed in Table 4. For the entire cohort, the observed number of SMs was 1.75-fold more than expected compared with the age-, sex-, and calendar year–matched Alberta general population (SIR, 1.75; 95% CI, 1.32-2.28; P < .01), leading to 4.3 excess cases of cancer per 1000 person years of follow-up. In those who received TBI, the observed number of SMs was 1.41-fold greater than expected (95% CI, 0.90-2.12; P = .07) vs 2.10-fold in those who did not receive TBI (95% CI, 1.45-2.96; P < .01), although, importantly, SIR calculations were not adjusted for covariates and time posttransplant (only total person years of follow-up). Younger age at transplant was associated with a greater risk of SM compared with the general population (SIR of 2.31 [95% CI, 1.48-3.43; P < .01] for those aged ≤50 years compared with SIR of 1.48 [95% CI, 1.02-2.10; P = .02] for those aged >50 years). Three other variables were found to be associated with a significant excess risk of SM compared with the general population: grades II to IV aGVHD (SIR, 2.78; 95% CI, 1.62-4.46; P < .01) with an EAR of 9.4, moderate to severe cGVHD (SIR, 2.28; 95% CI, 1.54-3.25; P < .01) with an EAR of 7.9, and a primary diagnosis of a lymphoid malignancy (SIR, 2.72; 95% CI, 1.70-4.12; P < .01) with an EAR of 13.8.
SIRs according to major sites of malignancy are detailed in supplemental Table 1. allo-HCT recipients in the cohort experienced significant excess risks of lung, oropharyngeal, and thyroid cancers, as well as melanoma (SIRs ranging from 2.67-7.10). Observed case numbers were too small to draw conclusions regarding the effect of TBI on excess risks of malignancy at each tumor site.
Discussion
The goal of the present study was to determine whether the addition of low-dose TBI (400 cGy) to a uniform myeloablative fludarabine and busulfan regimen is associated with an increased risk of SM in a predominantly adult allo-HCT population. In the entire cohort, we found a cumulative incidence of SM of 11.5% at 15 years’ post-HCT. Importantly, there did not seem to be a plateau in the cumulative incidence of SM after transplant. The cohort developed SMs at approximately double the rate of the age-, sex-, and calendar year–matched Alberta general population. However, in the multivariable model, low-dose TBI was not associated with SM. Rather, we found that number of pre-HCT cycles of chemotherapy (a 7% increase in risk for each cycle of therapy) and occurrence of cGVHD (although not statistically significant) were associated with SM. Important caveats to our study include that: (1) we studied a predominantly adult population; and (2) the follow-up of the TBI group was relatively short (8.2 years) with respect to development of SMs.
The risk of SM after allo-HCT has been well documented in the literature: consistent with our findings, the reported cumulative incidences range from 3% to 20% at 15 to 20 years’ post-HCT.1,8,17 -20 In addition, similar to our findings, an approximately twofold rate of development of SM in allo-HCT survivors compared with the general population has been described.1,8,17,18,21,22
The relationship between allo-HCT conditioned with TBI and SMs has been previously evaluated. TBI, especially when unfractionated, has been consistently reported as a risk factor for the development of SMs, particularly for skin, thyroid, breast, and liver cancers.1,5 -8,17 However, these studies have focused almost exclusively on the use of high-dose TBI regimens.1,8,22,23 In recent years, there has been increasing use of conditioning protocols that include lower doses of TBI (typically <600 cGy) with the goal of harnessing the antileukemic and immunosuppressive properties of TBI while reducing conditioning-related toxicity. For example, low-dose TBI has been used as part of non-myeloablative conditioning regimens,24,25 has been added to reduced-toxicity myeloablative conditioning regimens,9,13 and has been added to conditioning regimens for nonmalignant diseases when graft rejection is a concern.11,12 It is therefore important to understand whether low-dose TBI is associated with SM in the same fashion as high-dose TBI.
To our knowledge, only a single previous study has examined the risk of SM in those receiving allo-HCT with low-dose TBI as part of conditioning: Baker et al17 examined the association between TBI dose and SM in a large single-center analysis of almost 5000 patients. A key finding in this study was that, in contrast to high-dose TBI (≥600 cGy), low-dose TBI (200-450 cGy) was not associated with a higher risk of SM compared with chemotherapy-alone conditioning. Similarly, both those who received low-dose TBI conditioning and chemotherapy-alone conditioning experienced a twofold risk of SM compared with the general population, less than the three- to eightfold risk experienced by those who received high-dose TBI conditioning. Our findings are in concordance with those of Baker et al in that we found that low-dose TBI (400 cGy) was not associated with an increased risk of SM compared with chemotherapy-alone conditioning, yet both those who received low-dose TBI and those who received chemotherapy alone still experienced an elevated risk of SM compared with the general population. Our study, however, is unique and increases confidence in the finding that low-dose TBI does not add to risk of SM because all patients in our cohort, regardless of TBI, received uniform conditioning chemotherapy. In contrast, in the study by Baker et al, patients who received low-dose TBI did so in the context of non-myeloablative conditioning (fludarabine/TBI), whereas those who received chemotherapy-only regimens did so in the context of myeloablative conditioning; thus, the effect of differing chemotherapy intensity on risk of SM could not be controlled for. Furthermore, although Baker et al analyzed the association between low-dose TBI and SM in a non-myeloablative setting, we analyzed this association in a myeloablative setting, thus extending the published knowledge of the late effects of low-dose TBI across varying conditioning protocols.
An important secondary finding of our study is the significant contribution of pre-HCT chemotherapy to risk of SM after allo-HCT. To our knowledge, this study is the first to specifically examine the effect of pre-HCT chemotherapy, whether received for the treatment of the malignancy for which allo-HCT was undertaken or a prior malignancy, on risk of SM after allo-HCT. Although patients with lymphoid neoplasms, particularly CLL, are known to have an intrinsically higher risk of SM even in the absence of allo-HCT, these patients also tend to have an extensive treatment history before allo-HCT, complicating the assessment of risk factors for SM after allo-HCT.6,26,27 Our multivariable analysis suggests that the risk of SM is not necessarily driven by underlying diagnosis (lymphoma/CLL vs others) but by the extent of pre-HCT chemotherapy. Although the contribution of pre-HCT chemotherapies to the risk of SM after allo-HCT is a novel finding, it is perhaps not surprising given that non-HCT therapies for hematologic malignancies are known to be associated with SM.28-32 In contrast, we found that localized radiotherapy before HCT was not associated with SM: we hypothesize that this lack of association is due to the wide variety of anatomic locations, doses, and fractions of radiotherapy administered in this small subset of the cohort.
Our study was also able to comment on other variables associated with SM risk. In contrast to a subset of the prior literature,19,21,22 and in agreement with another subset of the literature,17,33 we found that, within our cohort, older age at time of transplant was not a significant risk factor for the development of SM. This discrepancy is likely due to variation in multivariable modeling methodology: typical Cox regression models use time since study entry as the time scale of the model, thus failing to account for the inherent age-associated increase in risk of malignancy.15 Rather, the use of patient age as the time scale allows for direct control of this follow-up age effect. In fact, compared with the age-, sex-, and calendar year–matched general population, younger patients in our cohort (aged ≤50 years at transplant) experienced a greater risk of SM (SIR, 2.31) compared with older patients (SIR, 1.48). This latter finding replicates the previous literature describing, compared with the general population, that younger allo-HCT recipients face a higher burden of excess malignancies compared with older allo-HCT recipients.1,17 This finding was not replicated in our multivariable analysis, perhaps due to the relatively small number of SMs in the cohort and relatively short follow-up. Finally, within the cohort, we found cGVHD to be associated with SM, although with borderline statistical significance. This borderline finding is likely due to the fact that cGVHD has been particularly associated with squamous cell carcinomas,1,8 and our analysis did not differentiate between underlying histology.
The current study has limitations. First, our relatively small study population limited the number of SMs that we observed. As a result, we were unable to analyze risk factors for specific SM sites and histology. Second, as in other retrospective studies of SMs after allo-HCT, we were unable to account for lifestyle (eg, tobacco use) and genetic predispositions that could have contributed to risk of SM. Nevertheless, we do not have reason to suspect that the distribution of these predispositions would have been significantly different between those who did and did not receive TBI. Third, our cohort did not include children: it is known that children may be more susceptible to radiation-related SMs.1 Thus, our results cannot be generalized to a pediatric population. Finally, although our median follow-up of the cohort was nearly 10 years, follow-up was slightly shorter in the TBI group, and it is possible that TBI will become a more important contributor to SM risk with further follow-up.
Our study has important strengths. First, we obtained SM data from a centralized public provincial cancer registry. Because all malignancies in the province are reported to the registry, we expect that we achieved near complete ascertainment of SMs. Second, our program continues to follow up patients indefinitely after allo-HCT at 1 of 2 major provincial cancer centers with a shared medical record, allowing accurate collection of transplant-related data and outcomes. Third, this single-province analysis allowed for a detailed review of pretransplant therapies which would not typically be available in large registry studies. Finally, because low-dose TBI was added to an unvarying standard conditioning chemotherapy protocol, we were able to study the effect of TBI without confounding by varying chemotherapy.
In summary, our findings suggest that allo-HCT conditioning regimens that include low-dose TBI are not associated with an increased risk of SM compared with chemotherapy-alone conditioning. However, allo-HCT recipients in our cohort still experience malignancies at a rate that is approximately double that of the general population, and there does not seem to be a plateau in SMs at 15 years’ post-transplant. These findings support the use of low-dose TBI as part of conditioning for allo-HCT, in which it may be an important tool in the maintenance of an antileukemic effect and/or in the reduction of the risk of graft rejection, while reducing conditioning-related toxicity. Furthermore, these findings will inform providers when counseling allo-HCT recipients regarding late toxicities and will inform the development of survivorship care plans as they pertain to screening for SMs. Specifically, we highlight the relevance of including pre-HCT chemotherapy exposures in the clinical risk assessment for SM after allo-HCT.
Acknowledgments
The authors thank their patients and the multidisciplinary staff of the Alberta Blood & Marrow Transplant Program.
Authorship
Contribution: L.N. contributed to study design, interpreted the data, and wrote the manuscript; T.A. analyzed data and reviewed the manuscript; S.N. and H.S. provided statistical support and reviewed the manuscript; S.B. collected data and reviewed the manuscript; L.S., A.D., M.S., P.D., A.C., and J.S. reviewed the manuscript and provided scientific input; and K.J. conceived the study idea and supervised execution of the project, designed the study, analyzed and interpreted data, and contributed to manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kareem Jamani, Room 603, South Tower, Foothills Medical Centre, 1403 29 St NW, Calgary, AB, T2N 2T9, Canada; e-mail: kareem.jamani@ahs.ca



