

Key Points
Elevated fibrinogen, in conjunction with accelerated formation of FXIIa, may promote compact fibrin clot architecture in COVID-19.
A dense fibrin network and dysregulated fibrinolysis collectively contribute to a high incidence of thrombotic events in COVID-19.

Abstract
The high incidence of thrombotic events suggests a possible role of the contact system pathway in COVID-19 pathology. In this study, we determined the altered levels of factor XII (FXII) and its activation products in critically ill patients with COVID-19 in comparison with patients with severe acute respiratory distress syndrome related to the influenza virus (acute respiratory distress syndrome [ARDS]-influenza). Compatible with those data, we found rapid consumption of FXII in COVID-19 but not in ARDS-influenza plasma. Interestingly, the lag phase in fibrin formation, triggered by the FXII activator kaolin, was not prolonged in COVID-19, as opposed to that in ARDS-influenza. Confocal and electron microscopy showed that increased FXII activation rate, in conjunction with elevated fibrinogen levels, triggered formation of fibrinolysis-resistant, compact clots with thin fibers and small pores in COVID-19. Accordingly, clot lysis was markedly impaired in COVID-19 as opposed to that in ARDS-influenza. Dysregulated fibrinolytic system, as evidenced by elevated levels of thrombin-activatable fibrinolysis inhibitor, tissue-plasminogen activator, and plasminogen activator inhibitor-1 in COVID-19 potentiated this effect. Analysis of lung tissue sections revealed widespread extra- and intravascular compact fibrin deposits in patients with COVID-19. A compact fibrin network structure and dysregulated fibrinolysis may collectively contribute to a high incidence of thrombotic events in COVID-19.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a multisystem disease emanating from the respiratory tract that has been designated coronavirus disease (COVID)-19.1-3 Rapidly accumulating data suggest that a major underlying molecular mechanism in COVID-19–related morbidity and mortality is widespread endothelial injury associated with hyperactivation of the immune system, leading to numerous hemostatic abnormalities.4-8 The clinical relevance of these processes is highlighted by the association between abnormal levels of D-dimer and the 28-day mortality rate in patients with COVID-199-13 and postmortem studies stressing the presence of microthrombi and capillarostasis in the lungs of affected subjects.14,15
The high incidence of thrombotic events (in particular, deep vein thrombosis and pulmonary embolism), in conjunction with mildly prolonged activated partial thromboplastin time,16,17 suggests that coagulation factor XII (FXII) has a role in COVID-19 coagulopathy. FXII is a serine protease of the contact-phase system of blood coagulation that circulates in plasma as a single-chain zymogen.18 After contact with anionic surfaces such as kaolin, but also extracellular RNA released from damaged cells,19 neutrophil extracellular traps (NETs),20 or polyphosphates secreted from activated platelets,21 FXII undergoes autoactivation to αFXIIa (herein referred to as FXIIa). FXIIa cleaves plasma prekallikrein (PK) to kallikrein (PKa), which reciprocally activates FXII and amplifies FXIIa generation. As a consequence, the plasma kallikrein-kinin system is activated, leading to the release of the vasodilatory and vascular barrier disrupting peptide bradykinin from high-molecular-weight kininogen (HK).22-25 Overall, activation of the contact-phase system may contribute to an increased production of thrombin and fibrin, although FXIIa/PKa-mediated conversion of plasminogen to plasmin may also affect fibrinolysis.26
A congenital deficiency of FXII in humans does not cause any bleeding complications, suggesting that FXII is dispensable for physiological hemostasis and fibrin formation.27 However, the contact-phase pathway may play an important role in the development of thrombosis when contact surfaces are exposed in scenarios such as trauma injury or bacterial and viral infections.27-29 Indeed, numerous animal studies have confirmed a critical function of FXII in thrombus growth and stabilization under the mentioned conditions and have provided the rationale for the development of new FXIIa inhibitors that ensure thromboprotection in patients without causing a bleeding complication.27-30
Given the high incidence of thromboembolic complications in severely ill patients with COVID-19,16,17 we investigated the contribution of FXII to clot formation and its architecture in our COVID-19 cohort in comparison with patients infected with the influenza virus.
Materials and methods
Additional procedures are described in the supplemental Methods.
Study population
Plasma samples of patients with COVID-19 were obtained from Charité-University Medicine, Berlin, Germany (Berlin cohort), and the Hanover Medical School, Hanover, Germany (Hanover cohort). Plasma samples from acute respiratory distress syndrome (ARDS) related to influenza were provided by the Hanover Medical School. All samples were obtained within 6 days after the onset of ARDS. All investigations were approved by the local ethics committees (Hanover samples: SEPSIS/ARDS Registry 8146_BO_K_2018; Berlin samples: EA2/066/20), and written informed consent was obtained from all participants or their next of kin. Patients with COVID-19 were classified as moderate (hospitalized, no invasive ventilation; WHO severity score: 3-4) or severe (high flow O2 or intubated and mechanically ventilated; WHO severity score: 5-7) as previously described.31 Donor (healthy subjects) samples were provided by Charité-University Medicine, Berlin, Germany (EA2/075/15), and Justus-Liebig University, Giessen, Germany (05/00). Blood was collected into sodium citrate–containing blood specimen tubes via standard venipuncture. All blood biospecimens were processed without a stabilizing reagent, at room temperature, within 5 hours of collection, and stored at −80°C. All plasmas were used in analyses of circulating proteins as well as coagulation and fibrinolytic activity except when insufficient plasma from an individual subject was available. Baseline demographics and clinical characteristics of the donors and patients are shown in Table 1.
Baseline demographics and clinical characteristics of donors and patients with COVID-19 or ARDS-influenza who provided plasma samples
| Hanover cohort | Berlin cohort | |||||
|---|---|---|---|---|---|---|
| ARDS-influenza | COVID-19 (WHO 5-7) | Donor | COVID-19 (WHO 5-7) | COVID-19 (WHO 3-4) | Donor | |
| Patients, n | 25 | 21 | 21 | 15 | 15 | 15 |
| Age, y [range] | 56 [20-86] | 59 [19-82] | 60 [20-79] | 61 [22-84] | 61 [26-80] | 61 [24-82] |
| Sex, male, % | 87 | 90 | 88 | 69 | 67 | 68 |
| BMI [range], kg/m2 | 25 [20-36] | 29 [15-62] | 26 [19-45] | 29 [25-36] | 24 [20-36] | 25 [22-37] |
| 65 score | ||||||
| 0 | 0 | 0 | — | NA | NA | — |
| 1 | 3 | 2 | — | NA | NA | — |
| 2 | 5 | 6 | — | NA | NA | — |
| 3 | 12 | 9 | — | NA | NA | — |
| 4 | 5 | 4 | — | NA | NA | — |
| 28-d mortality, % | 30 | 14.3 | — | 8 | 0 | — |
| LOS ICU, d [range] | 19 [6-73] | 27 [3-63] | — | 30 [5-220] | NA | — |
| Ventilation, d [range] | 15 [3-66] | 16 [4-50] | — | 26 [5-220] | NA | — |
| ECMO, % | 30 | 24 | — | 46 | NA | — |
| SOFA score [range] | 10 [5-16] | 13 [9-17] | — | 10 [2-12] | NA | — |
| CRP, mg/L [range] | 264 [31-406] | 151 [68-292] | — | 85 [27-411] | 29 [1-148] | — |
| Leukocytes, ×109/L [range] | 16 [22-90] | 9 [4-36] | — | 10 [5-27] | 7 [4-22] | — |
| Platelets, ×109/L [range] | 199 [70-653] | 247 [99-581] | — | 286 [129-635] | 334 [173-602] | — |
| Lactate, mM [range] | 1.3 [0.7-4.8] | 1.8 [0.7-5.6] | — | 1.7 [0.4-6.6] | 1.3 [1.0-2.6] | — |
| Procalcitonin, µg/L [range] | 1.7 [0.2-79.5] | 0.6 [0.1-66.1] | — | 0.6 [0.1-25] | 0.1 [0-1] | — |
| d-dimer, mg/L [range] | NA | 4 [1-35] | — | NA | NA | — |
| Fibrinogen, g/L [range] | 4.0 [3.2-9.0] | 8.0 [4.0-9.0] | — | NA | NA | — |
| Anticoagulant | ||||||
| Heparin, n | 15 | 14 | — | 8 | 7 | — |
| PTT, s [range] | 38 [25-59] | 36 [26-55] | — | 41 [30-67] | 34 [30-60] | — |
| Hanover cohort | Berlin cohort | |||||
|---|---|---|---|---|---|---|
| ARDS-influenza | COVID-19 (WHO 5-7) | Donor | COVID-19 (WHO 5-7) | COVID-19 (WHO 3-4) | Donor | |
| Patients, n | 25 | 21 | 21 | 15 | 15 | 15 |
| Age, y [range] | 56 [20-86] | 59 [19-82] | 60 [20-79] | 61 [22-84] | 61 [26-80] | 61 [24-82] |
| Sex, male, % | 87 | 90 | 88 | 69 | 67 | 68 |
| BMI [range], kg/m2 | 25 [20-36] | 29 [15-62] | 26 [19-45] | 29 [25-36] | 24 [20-36] | 25 [22-37] |
| 65 score | ||||||
| 0 | 0 | 0 | — | NA | NA | — |
| 1 | 3 | 2 | — | NA | NA | — |
| 2 | 5 | 6 | — | NA | NA | — |
| 3 | 12 | 9 | — | NA | NA | — |
| 4 | 5 | 4 | — | NA | NA | — |
| 28-d mortality, % | 30 | 14.3 | — | 8 | 0 | — |
| LOS ICU, d [range] | 19 [6-73] | 27 [3-63] | — | 30 [5-220] | NA | — |
| Ventilation, d [range] | 15 [3-66] | 16 [4-50] | — | 26 [5-220] | NA | — |
| ECMO, % | 30 | 24 | — | 46 | NA | — |
| SOFA score [range] | 10 [5-16] | 13 [9-17] | — | 10 [2-12] | NA | — |
| CRP, mg/L [range] | 264 [31-406] | 151 [68-292] | — | 85 [27-411] | 29 [1-148] | — |
| Leukocytes, ×109/L [range] | 16 [22-90] | 9 [4-36] | — | 10 [5-27] | 7 [4-22] | — |
| Platelets, ×109/L [range] | 199 [70-653] | 247 [99-581] | — | 286 [129-635] | 334 [173-602] | — |
| Lactate, mM [range] | 1.3 [0.7-4.8] | 1.8 [0.7-5.6] | — | 1.7 [0.4-6.6] | 1.3 [1.0-2.6] | — |
| Procalcitonin, µg/L [range] | 1.7 [0.2-79.5] | 0.6 [0.1-66.1] | — | 0.6 [0.1-25] | 0.1 [0-1] | — |
| d-dimer, mg/L [range] | NA | 4 [1-35] | — | NA | NA | — |
| Fibrinogen, g/L [range] | 4.0 [3.2-9.0] | 8.0 [4.0-9.0] | — | NA | NA | — |
| Anticoagulant | ||||||
| Heparin, n | 15 | 14 | — | 8 | 7 | — |
| PTT, s [range] | 38 [25-59] | 36 [26-55] | — | 41 [30-67] | 34 [30-60] | — |
BMI, body mass index; CRB 65, confusion, respiratory rate, blood pressure, age 65 score; LOS ICU, length of intensive care unit stay; ECMO, extracorporeal membrane oxygenation; SOFA, sequential organ failure assessment; PTT, partial thromboplastin time.
Lung specimens were obtained from 10 patients with ARDS (5 COVID-19, 5 influenza) and 5 donors at autopsy. Time from death to autopsy was matched between donors and patients. All investigations were approved by the local ethics committees (Medical Faculty of Justus-Liebig University: 29/01 and Medical University of Graz: 32-362ex19/20), and written informed consent was obtained from all participants or their next of kin, if required. Baseline demographics and clinical characteristics of lung tissue donors are shown in Table 2.
Baseline demographics and clinical characteristics of donors and patients with COVID-19 or ARDS-influenza who provided lung tissue specimens at autopsy
| Patient | Age, y | Sex | Background | Ventilation, d | Anticoagulant |
|---|---|---|---|---|---|
| COVID-19 | |||||
| 1 | 82 | Male | Diffuse alveolar damage | 0 | Heparin |
| 2 | 77 | Male | Diffuse alveolar damage | 2 | Heparin |
| 3 | 72 | Male | Diffuse alveolar damage | 6 | Heparin |
| 4 | 65 | Male | Diffuse alveolar damage | 33 | Heparin |
| 5 | 79 | Female | Diffuse alveolar damage | 2 | Heparin |
| ARDS-influenza | |||||
| 1 | 67 | Male | CAP | 6 | Heparin |
| 2 | 72 | Male | CAP | 10 | Heparin |
| 3 | 77 | Male | CAP | 5 | Heparin |
| 4 | 81 | Female | CAP | 10 | Heparin |
| 5 | 80 | Female | CAP | 3 | Heparin |
| Donor | |||||
| 1 | 82 | Female | Recurrent myocardial infarction | 0 | No |
| 2 | 75 | Female | Heart and lung failure | 0 | Heparin |
| 3 | 64 | Male | Myocardial infarction | 0 | Heparin |
| 4 | 77 | Female | Dilated cardiomyopathy (right) | 0 | Heparin |
| 5 | 75 | Male | Dilated cardiomyopathy (right) | 0 | Heparin |
| Patient | Age, y | Sex | Background | Ventilation, d | Anticoagulant |
|---|---|---|---|---|---|
| COVID-19 | |||||
| 1 | 82 | Male | Diffuse alveolar damage | 0 | Heparin |
| 2 | 77 | Male | Diffuse alveolar damage | 2 | Heparin |
| 3 | 72 | Male | Diffuse alveolar damage | 6 | Heparin |
| 4 | 65 | Male | Diffuse alveolar damage | 33 | Heparin |
| 5 | 79 | Female | Diffuse alveolar damage | 2 | Heparin |
| ARDS-influenza | |||||
| 1 | 67 | Male | CAP | 6 | Heparin |
| 2 | 72 | Male | CAP | 10 | Heparin |
| 3 | 77 | Male | CAP | 5 | Heparin |
| 4 | 81 | Female | CAP | 10 | Heparin |
| 5 | 80 | Female | CAP | 3 | Heparin |
| Donor | |||||
| 1 | 82 | Female | Recurrent myocardial infarction | 0 | No |
| 2 | 75 | Female | Heart and lung failure | 0 | Heparin |
| 3 | 64 | Male | Myocardial infarction | 0 | Heparin |
| 4 | 77 | Female | Dilated cardiomyopathy (right) | 0 | Heparin |
| 5 | 75 | Male | Dilated cardiomyopathy (right) | 0 | Heparin |
CAP, community-acquired pneumonia.
Plasma clot formation and lysis
Twenty microliters of plasma were preincubated for 10 minutes with 20 mL of 0.1 M imidazole buffer (pH 7.4) and 20 mL of 0.3 mg/mL kaolin in a clear, flat-bottomed, 96-well plate. Clotting was initiated by the addition of 20 mL of 20 mM CaCl2 in the absence or presence of tissue plasminogen activator (tPA) (25 ng/mL final; Sekisui Diagnostics, Burlington, MA). Turbidity was monitored at 405 nm (A405) every 30 seconds for 60 minutes at 37°C using a SpectraMax 190 (Molecular Devices, Biberach, Germany). In some experiments, COVID-19 plasma was preincubated with hirudin (5 IE/mL final; Diapharma, West Chester, OH), and the clotting was induced by batroxobin (5 U/mL final; Enzyme Research Laboratories, South Bend, IN).
Fibrin formation and lysis in a purified system
Thrombin (5 nM final; Sekisui Diagnostics) was mixed with fibrinogen (2-9 g/L final; ThermoFisher Scientific, Waltham, MA), preincubated with either FXII or FXIIa (10-40 μg/mL final; both from Sekisui Diagnostics) in a total volume of 25 μL of 0.1 M imidazole buffer in a clear, flat-bottomed, 96-well plate. Fibrin formation was initiated by the addition of 20 μL of 20 mM CaCl2. To measure fibrinolysis, tPA (0.1 µg/mL final) and plasminogen (20 µg/mL final; Enzyme Research Laboratories) were added to the clotting solution. Turbidity was monitored as described earlier. In some experiments, FXIIa (40 μg/mL final) was preincubated with corn trypsin inhibitor (CTI; 0.01 U/mL final; Sekisui Diagnostics) before mixing with fibrinogen. FXII and FXIII contaminations were not detected in fibrinogen preparation by means of western blot analysis and enzyme-linked immunosorbent assay.
FXII decay in plasma
Endogenous FXII was depleted from plasma using the goat anti-FXII antibody (cat. no. 206-0056; Zytomed Systems) covalently attached to magnetic beads (ThermoFisher Scientific). Afterward, 100 µL of plasma was supplemented with 30 nM exogenous FXII, and the sample was incubated for 1 hour at 37°C. Aliquots were withdrawn after the indicated time points and analyzed by western blot analysis. In some experiments, plasma was preincubated with 12 mg/mL CTI 30 minutes before the addition of FXII.
Activity assays
Statistics
Statistical analysis was performed in the ggpubr package of R (version 4).34,35 Data are expressed as single data points with boxplot overlay indicating median and interquartile range, unless indicated otherwise. Multiple groups were compared by nonparametric Kruskal-Wallis test. Correlations were performed using Spearman’s rank correlation coefficient.
Results
FXII is activated in patients with severe COVID-19
In the Berlin cohort, the plasma levels of FXII were decreased in patients with severe COVID-19, as compared with donors (Figure 1A-B; moderate: WHO severity score, 3-4; severe: WHO severity score, 5-7). Disappearance of the FXII in plasma typically corresponds to its activation and conversion into the 2-chain FXIIa protein composed of the 50-kDa heavy chain and 30-kDa light chain. Detection of FXIIa in plasma is, however, hindered by its rapid inactivation and complex formation with C1 esterase inhibitor (C1INH). Thus, to better monitor the presence of FXIIa in COVID-19 plasma, we monitored products of its activation, such as cleaved HK and PKa. As expected, disappearance of FXII in plasma was accompanied by HK cleavage, seen as diminished signal intensity of the intact HK band at 130 kDa (Figure 1C-D). A decrease in intact HK levels was associated with the appearance of cleaved HK fragments: the cleaved HK light chain band migrating at 55 kDa and an additional 45-kDa band representing a degradation product of the 55-kDa cleaved HK light chain. To further examine whether the reduction in intact levels of FXII and HK is a result of the contact system activation, we measured the activity of plasma PKa. PKa-like activity was markedly elevated in patients with severe COVID-19 in comparison with donors and patients with moderate SARS-CoV-2 infection (Figure 1E). Furthermore, a strong negative correlation between the intact HK/albumin ratio and the PKa-like activity was observed in the plasma of patients with severe COVID-19 (Figure 1F). Purified plasma proteins and deficient plasma samples were used to demonstrate the specificity of the bands shown in western blots (Figure 1A,C; right).

Activation of the contact-phase system in plasma of patients critically ill with COVID-19. (A,C) Western blot analysis (left) of FXII (A) and HK (C) in plasma from patients with moderate or severe COVID-19 (SARS-CoV-2) and donors (healthy subjects). Four of 15 patients with moderate or severe COVID-19 and 3 of 15 donors are represented. Albumin was used as the loading control. The specificity of the antibodies used is shown (right). (B,D) Densitometric analysis of panels A and C, respectively. COVID-19 moderate/severe, n = 15; donor n = 15. (E) PKa-like activity in plasma of patients with moderate (n = 14) or severe (n = 14) COVID-19 and donors (n = 15). (F) Correlation between the levels of intact HK and PKa-like activity in plasma of patients with severe COVID-19 (n = 14). Correlation was performed according to Spearman’s rank correlation coefficient. *P < .05; **P < .01; ***P < .001.
Activation of the contact-phase system in plasma of patients critically ill with COVID-19. (A,C) Western blot analysis (left) of FXII (A) and HK (C) in plasma from patients with moderate or severe COVID-19 (SARS-CoV-2) and donors (healthy subjects). Four of 15 patients with moderate or severe COVID-19 and 3 of 15 donors are represented. Albumin was used as the loading control. The specificity of the antibodies used is shown (right). (B,D) Densitometric analysis of panels A and C, respectively. COVID-19 moderate/severe, n = 15; donor n = 15. (E) PKa-like activity in plasma of patients with moderate (n = 14) or severe (n = 14) COVID-19 and donors (n = 15). (F) Correlation between the levels of intact HK and PKa-like activity in plasma of patients with severe COVID-19 (n = 14). Correlation was performed according to Spearman’s rank correlation coefficient. *P < .05; **P < .01; ***P < .001.
Fibrinogen and FXIIa regulate fibrin network density in COVID-19
To assess whether enhanced activation of FXII in critically ill patients with COVID-19 represents a characteristic feature of SARS-CoV-2 infection, we analyzed plasma samples of patients with ARDS related to influenza virus infection. The decrease in FXII plasma levels in severe COVID-19 in comparison with that in donors was confirmed in the independent (Hanover) cohort (Hanover cohort) of patients (Figure 2A). Furthermore, the levels of FXII in COVID-19 were significantly lower than those in ARDS-influenza (Figure 2B). More surprising, the lag phase in fibrin formation, triggered by the FXII activator kaolin, was shorter in plasma of patients with COVID-19 compared with patients with ARDS-influenza (Figure 2C). High plasma levels of FVIII:C in COVID-19 as opposed to ARDS-influenza provide a possible explanation for this effect (Figure 2D). Notably, patients with COVID-19 or ARDS-influenza received the same daily dose of unfractionated heparin, excluding iatrogenic anticoagulation as a cause of prolonged lag phase in fibrin formation in ARDS-influenza. In addition, we excluded lupus anticoagulant and the presence of anti-FXII antibodies as a cause of FXII deficiency in patients with COVID-19 in our cohort (data not shown).
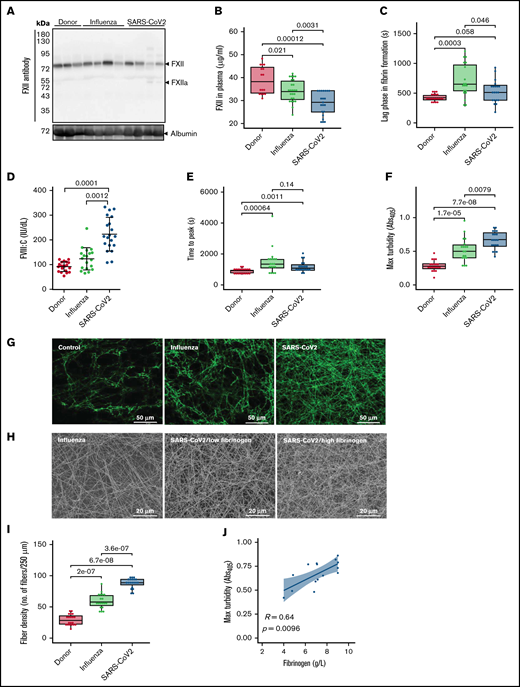
Formation of dense fibrin clots in plasma of patients with severe COVID-19. (A) Western blot analysis of factor XII in plasma of patients with ARDS-influenza (Influenza) or COVID-19 (SARS-CoV-2), as well as donors. Data from 4 of 21 patients with COVID-19, 4 of 25 patients with ARDS-influenza, and 3 of 21 donors are shown. Albumin was used as the loading control. (B) FXII levels in plasma of patients with ARDS-influenza (n = 25) or COVID-19 (n = 21) and donors (n = 16) as assessed by enzyme-linked immunosorbent assay. (C) Lag phase in fibrin formation triggered by kaolin. Influenza, n = 19; SARS-CoV-2, n = 20; donor, n = 20. (D) FVIII activity (FVIII:C) in plasma of patients and donors. Influenza, n = 19; SARS-CoV-2, n = 20; donor, n = 20. (E-F) Time to reach the turbidity peak (E) and maximum (Max) turbidity (F) values for influenza (n = 19), SARS-CoV-2 (n = 20), and donor (n = 20) plasma. Clot formation was induced by the addition of kaolin to plasma. (G) Representative laser scanning confocal microscopy images of fibrin fibers in clots formed from influenza (n = 19), SARS-CoV-2 (n = 20), and donor (n = 20) plasma. Fibrin fibers were stained with an anti-fibrinogen/fibrin antibody followed by an Alexa Fluor 488-conjugated secondary antibody. (H) Representative scanning electron microscopy images of fibrin network in clots generated from influenza plasma (n = 5) as well as low- and high-fibrinogen SARS-CoV-2 (n = 5 per group) plasma. (I) Fibrin fiber density in donor (n = 20), ARDS-influenza (n = 19), and COVID-19 (n = 20) clots. From each patient, 3 separate clots were prepared, 5 images were taken in different areas of the clots, and fibril density was determined in all images. (J) Correlation between maximum turbidity values and fibrinogen levels in plasma of patients with COVID-19 (n = 15; those patients with available fibrinogen levels were included into the analysis). Correlation was performed according to Spearman’s rank correlation coefficient.
Formation of dense fibrin clots in plasma of patients with severe COVID-19. (A) Western blot analysis of factor XII in plasma of patients with ARDS-influenza (Influenza) or COVID-19 (SARS-CoV-2), as well as donors. Data from 4 of 21 patients with COVID-19, 4 of 25 patients with ARDS-influenza, and 3 of 21 donors are shown. Albumin was used as the loading control. (B) FXII levels in plasma of patients with ARDS-influenza (n = 25) or COVID-19 (n = 21) and donors (n = 16) as assessed by enzyme-linked immunosorbent assay. (C) Lag phase in fibrin formation triggered by kaolin. Influenza, n = 19; SARS-CoV-2, n = 20; donor, n = 20. (D) FVIII activity (FVIII:C) in plasma of patients and donors. Influenza, n = 19; SARS-CoV-2, n = 20; donor, n = 20. (E-F) Time to reach the turbidity peak (E) and maximum (Max) turbidity (F) values for influenza (n = 19), SARS-CoV-2 (n = 20), and donor (n = 20) plasma. Clot formation was induced by the addition of kaolin to plasma. (G) Representative laser scanning confocal microscopy images of fibrin fibers in clots formed from influenza (n = 19), SARS-CoV-2 (n = 20), and donor (n = 20) plasma. Fibrin fibers were stained with an anti-fibrinogen/fibrin antibody followed by an Alexa Fluor 488-conjugated secondary antibody. (H) Representative scanning electron microscopy images of fibrin network in clots generated from influenza plasma (n = 5) as well as low- and high-fibrinogen SARS-CoV-2 (n = 5 per group) plasma. (I) Fibrin fiber density in donor (n = 20), ARDS-influenza (n = 19), and COVID-19 (n = 20) clots. From each patient, 3 separate clots were prepared, 5 images were taken in different areas of the clots, and fibril density was determined in all images. (J) Correlation between maximum turbidity values and fibrinogen levels in plasma of patients with COVID-19 (n = 15; those patients with available fibrinogen levels were included into the analysis). Correlation was performed according to Spearman’s rank correlation coefficient.
Further analysis of kaolin-triggered plasma clotting time revealed an increase in the time to reach the turbidity peak in both patient groups compared with donors, but no difference between ARDS-influenza and COVID-19 (Figure 2E). The density of the clot (indicated by the maximum turbidity measurement) was higher in both patient groups as opposed to donors. A direct comparison between clots of ARDS-influenza and COVID-19 showed significantly higher maximum turbidity values in the latter group (Figure 2F). Visualization of fibrin clots by laser scanning confocal microscopy and scanning electron microscopy revealed an increase in fibrin structure compactness with thinner fibers and smaller pores in clots from COVID-19 plasma, as compared with clots generated from ARDS-influenza plasma (Figure 2G-I). A detailed analysis of the clots generated from plasma of patients with COVID-19 demonstrated association between packing density of fibrin fibers and plasma fibrinogen concentration, with dense fibrin network in clots formed in plasma of patients exhibiting high fibrinogen levels (Figure 2H). A strong positive correlation was noted between maximum turbidity values and fibrinogen concentration in plasma of patients with COVID-19 (Figure 2J).
As the architecture of fibrin clots may be influenced not only by fibrinogen but also by FXIIa,36,37 we next analyzed the impact of these 2 proteins on the clot structure in a purified system. High concentrations of fibrinogen increased peak turbidity values, and this effect was potentiated by the addition of FXIIa (Figure 3A). Accordingly, CTI, the inhibitor of FXIIa, reduced the maximum turbidity of the clots generated by mixing fibrinogen and FXIIa (Figure 3B).
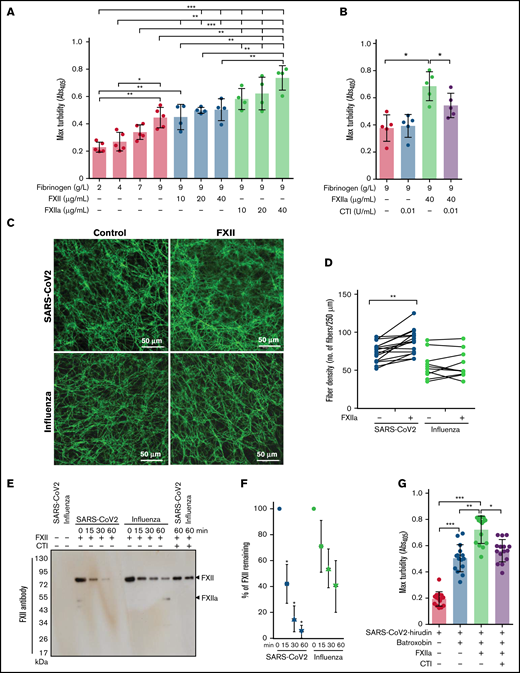
Impact of FXIIa on fibrin clot structure in plasma from patients with severe COVID-19. (A-B) Maximum turbidity values of fibrin clots generated in the purified system from increasing concentrations of fibrinogen and/or FXII/FXIIa, in the absence or presence of CTI. Clot formation was induced by thrombin (n = 4-5). (C) Representative laser scanning confocal microscopy images of fibrin fibers in clots formed from FXII-depleted SARS-CoV-2 or influenza plasma supplemented with FXII. Fibrin fibers were stained with an anti-fibrinogen/fibrin antibody followed by an Alexa Fluor 488-conjugated secondary antibody. (D) Fibrin fiber density in ARDS-influenza (n = 10) and COVID-19 (n = 10) clots generated in panel C. Per patient, 3 separate clots were prepared, 5 images were taken in different areas of the clots, and fibril density was determined in all images. Interconnections of paired data are shown. (E) Rate of FXII autoactivation in ARDS-influenza and SARS-CoV-2 plasma. FXII was added to FXII-depleted plasma, and its decay was monitored by Western blot assay, with an antibody directed against FXII. A representative blot is shown. (F) Quantification of FXII decay in ARDS-influenza and SARS-CoV-2 plasma in panel E. FXII signal at time point 0 was considered to be 100% (n = 20 per group). (G) Maximum turbidity values of fibrin clots generated by the addition of batroxobin to hirudin-preincubated plasma in the presence of FXIIa and/or CTI (n = 15 biological replicates). *P < .05; **P < .01; ***P < .001.
Impact of FXIIa on fibrin clot structure in plasma from patients with severe COVID-19. (A-B) Maximum turbidity values of fibrin clots generated in the purified system from increasing concentrations of fibrinogen and/or FXII/FXIIa, in the absence or presence of CTI. Clot formation was induced by thrombin (n = 4-5). (C) Representative laser scanning confocal microscopy images of fibrin fibers in clots formed from FXII-depleted SARS-CoV-2 or influenza plasma supplemented with FXII. Fibrin fibers were stained with an anti-fibrinogen/fibrin antibody followed by an Alexa Fluor 488-conjugated secondary antibody. (D) Fibrin fiber density in ARDS-influenza (n = 10) and COVID-19 (n = 10) clots generated in panel C. Per patient, 3 separate clots were prepared, 5 images were taken in different areas of the clots, and fibril density was determined in all images. Interconnections of paired data are shown. (E) Rate of FXII autoactivation in ARDS-influenza and SARS-CoV-2 plasma. FXII was added to FXII-depleted plasma, and its decay was monitored by Western blot assay, with an antibody directed against FXII. A representative blot is shown. (F) Quantification of FXII decay in ARDS-influenza and SARS-CoV-2 plasma in panel E. FXII signal at time point 0 was considered to be 100% (n = 20 per group). (G) Maximum turbidity values of fibrin clots generated by the addition of batroxobin to hirudin-preincubated plasma in the presence of FXIIa and/or CTI (n = 15 biological replicates). *P < .05; **P < .01; ***P < .001.
Because sustained activation of FXII was described in COVID-19,38 we next investigated the potential contribution of FXIIa to the regulation of fibrin clot structure in this patient group. To this end, FXII-depleted COVID-19 and ARDS-influenza plasma samples were recalcified in the absence or presence of exogenous FXII, and the fibrin clots were visualized by using the antibody against fibrinogen/fibrin. FXII increased fibrin network density but not fibrin fiber diameter in COVID-19 plasma (Figure 3C-D). Yet, no apparent effect of FXII on the clot architecture in ARDS-influenza was observed (Figure 3C-D). Interestingly, the most prominent effect of FXII on fibrin network density was observed in COVID-19 plasma samples containing high levels of fibrinogen (Figure 3D). Together, these results imply that COVID-19 plasma contains FXII autoactivation cofactor(s) that trigger generation of FXIIa. FXIIa then affects the fibrin clot structure in a thrombin-dependent and/or thrombin-independent manner. In line with this assumption, rapid decay of exogenous FXII in COVID-19 plasma was observed (Figure 3E-F). CTI markedly delayed disappearance of FXII suggesting that autoactivation of FXIIa occurred in COVID-19 and ARDS-influenza plasma samples (Figure 3E). To demonstrate a direct, thrombin-independent effect of FXIIa on the fibrin clot structure, we clotted hirudin-preincubated COVID-19 plasma with batroxobin in the presence of FXIIa and/or CTI and measured maximum turbidity. As shown in Figure 3G, FXIIa increased maximum turbidity, and this effect was diminished by CTI. These results are in line with the experiments performed in the purified system (Figure 3A-B). In sum, our results imply that FXIIa, in addition to its possible effect on thrombin generation, may also directly contribute to the fibrin network structure in COVID-19 plasma.
Elevated fibrin network density correlates with increased clot resistance to fibrinolysis
Compact fibrin network density has been found to impair clot fibrinolysis.39 Accordingly, we next evaluated the lysis resistance of fibrin clots in patient plasma, using an in vitro turbidimetric clot-lysis assay. Kaolin and tPA were added to plasma to initiate the intrinsic pathway of coagulation, followed by fibrin-dependent plasmin generation via tPA-mediated activation of plasminogen in the same sample. Whereas in donor plasma, the characteristic bell-shaped clot-lysis curve, representing the complete fibrin clot dissolution, was observed, only partial clot-lysis was detected in ARDS-influenza samples, and clot-lysis was absent in COVID-19 samples over the entire period of the experiment (Figure 4A). This observation is supported by the highest turbidity values at 60 minutes in COVID-19 samples (Figure 4B). Overall, clot lysis (as assessed by turbidity values at 60 minutes) was observed in 84% of patients with ARDS-influenza and only 30% of patients with COVID-19, suggesting pronounced deregulation of the fibrinolytic system in SARS-CoV-2–infected subjects in our cohort. As expected, increasing amounts of fibrinogen and FXIIa prolonged clot lysis time, with an additive effect being observed at the highest concentrations of both proteins (Figure 4C). The addition of CTI to the assay shortened clot lysis time (Figure 4D).
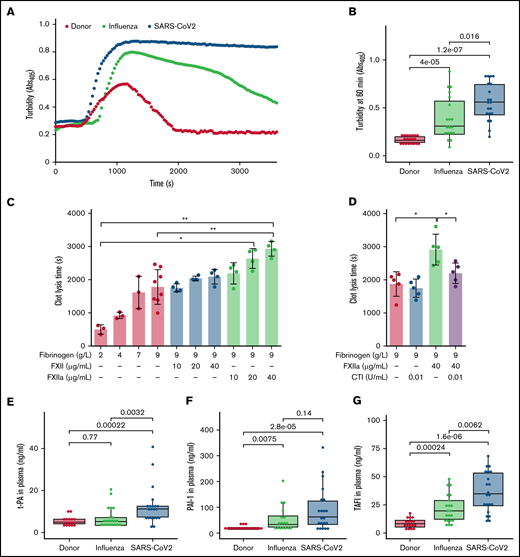
Dysregulated fibrinolysis in severe COVID-19. (A) Turbidimetric analysis of clot lysis in severe COVID-19, ARDS-influenza, and donor plasma. Representative clot lysis curves are shown. SARS-CoV-2, n = 20; ARDS-influenza; n = 19, donors, n = 20. (B) Turbidity values of the fibrin clots at 60 minutes. SARS-CoV-2, n = 20; ARDS-influenza, n = 19; donors, n = 20. (C-D) Clot lysis time. Clots were generated in a purified system with increasing concentrations of fibrinogen and/or FXII/FXIIa. Clot formation was induced by thrombin and clot lysis by plasmin generated from plasminogen by tPA. In some experiments, FXII was preincubated with CTI. Clot formation and lysis were monitored via turbidimetry (n = 3-5). (E-G) tPA (E), plasminogen activator inhibitor-1 (PAI-1; F), and thrombin-activatable fibrinolysis inhibitor (TAFI; G) levels in plasma of patients with COVID-19 (n = 21) or ARDS-influenza (n = 21) and donors (n = 17), as assessed by enzyme-linked immunosorbent assay. *P < .05; **P < .01.
Dysregulated fibrinolysis in severe COVID-19. (A) Turbidimetric analysis of clot lysis in severe COVID-19, ARDS-influenza, and donor plasma. Representative clot lysis curves are shown. SARS-CoV-2, n = 20; ARDS-influenza; n = 19, donors, n = 20. (B) Turbidity values of the fibrin clots at 60 minutes. SARS-CoV-2, n = 20; ARDS-influenza, n = 19; donors, n = 20. (C-D) Clot lysis time. Clots were generated in a purified system with increasing concentrations of fibrinogen and/or FXII/FXIIa. Clot formation was induced by thrombin and clot lysis by plasmin generated from plasminogen by tPA. In some experiments, FXII was preincubated with CTI. Clot formation and lysis were monitored via turbidimetry (n = 3-5). (E-G) tPA (E), plasminogen activator inhibitor-1 (PAI-1; F), and thrombin-activatable fibrinolysis inhibitor (TAFI; G) levels in plasma of patients with COVID-19 (n = 21) or ARDS-influenza (n = 21) and donors (n = 17), as assessed by enzyme-linked immunosorbent assay. *P < .05; **P < .01.
To test whether other components of the fibrinolytic system, such as tPA, PAI-1, and TAFI, may be dysregulated in patients critically ill with COVID-19, we measured their levels by means of enzyme-linked immunosorbent assay. The concentration of tPA was elevated in COVID-19, as compared with donors and patients with ARDS-influenza (Figure 4E). An increase of PAI-1 was also noted in the plasma of patients with ARDS-influenza or COVID-19, as opposed to that of donors, yet a significant difference between both patient groups was not detected (Figure 4F). Interestingly, TAFI was not only markedly elevated in both patient groups, compared with donors, but it was also higher in patients with COVID-19, compared with ARDS-influenza (Figure 4G).
Dense fibrin clots are observed in the lungs of patients with severe COVID-19
To demonstrate the in vivo relevance of our findings, we stained for fibrin the tissue sections obtained at autopsy from patients with SARS-CoV-2 or ARDS-influenza, as well as subjects who died of no respiratory cause. Notably, time from death to autopsy was matched for all groups examined. As demonstrated in Figure 5A, intra- and extravascular fibrin aggregates were observed in patients with severe COVID-19 or ARDS-influenza. However, in contrast to patients with ARDS-influenza, the deposits of fibrin in the lungs of those with COVID-19 appeared to be more widespread and evenly present, not only in vascular but also in alveolar spaces. In patients with ARDS-influenza, fibrin deposits were predominantly observed in alveolar spaces and in selected regions of the lung (Figure 5A,C). Overall, in COVID-19 lung tissue, fibrin clots were more compact and homogeneous whereas in ARDS-influenza lung tissue they were widespread and characterized by regions of high and low fibrin fiber density (Figure 5B,D).

High abundance of fibrin deposits in the lungs of patients with severe COVID-19. (A-B) Fibrin (red) accumulation in postmortem lung tissue sections of patients with severe COVID-19 or ARDS-influenza and donors (n = 5/group). Time from death to autopsy was matched for all groups examined. Bar represents 100 µm. (B) Randomly chosen high-magnification images of the COVID-19 and ARDS-influenza clots presented in panel A. Arrows indicate fibrin deposits. All patients available are represented. Fibrin fibers were stained with an anti-fibrinogen/fibrin antibody and then developed by incubation with fast red dye. (C) Fibrin abundance in COVID-19, ARDS-influenza, and donor lungs. From each patient, 5 images were obtained in different areas of the lung, and the percentage of total area was determined in all images (n = 5 per group). *P < .05; **P < .01. (D) Fibrin fiber density in COVID-19 and ARDS-influenza lungs. From each patient, 5 images of fibrin deposits were taken, and fibril density was determined in all of them (n = 5 per group).
High abundance of fibrin deposits in the lungs of patients with severe COVID-19. (A-B) Fibrin (red) accumulation in postmortem lung tissue sections of patients with severe COVID-19 or ARDS-influenza and donors (n = 5/group). Time from death to autopsy was matched for all groups examined. Bar represents 100 µm. (B) Randomly chosen high-magnification images of the COVID-19 and ARDS-influenza clots presented in panel A. Arrows indicate fibrin deposits. All patients available are represented. Fibrin fibers were stained with an anti-fibrinogen/fibrin antibody and then developed by incubation with fast red dye. (C) Fibrin abundance in COVID-19, ARDS-influenza, and donor lungs. From each patient, 5 images were obtained in different areas of the lung, and the percentage of total area was determined in all images (n = 5 per group). *P < .05; **P < .01. (D) Fibrin fiber density in COVID-19 and ARDS-influenza lungs. From each patient, 5 images of fibrin deposits were taken, and fibril density was determined in all of them (n = 5 per group).
Discussion
Many patients with severe COVID-19 exhibit coagulation abnormalities that mimic other systemic coagulopathies associated with severe infections, such as disseminated intravascular coagulation (DIC) and thrombotic microangiopathy.40 A high incidence of venous thromboembolism, pulmonary embolism, deep vein thrombosis, and multiple organ failure with a poor prognosis and outcome appears to be causally related to dysregulation of blood coagulation in critically ill patients with COVID-19. Besides an elevated inflammatory status that may induce monocyte-related coagulation and suppression of anticoagulant pathways, typical laboratory findings in patients with COVID-19 are increased D-dimer levels and elevated fibrinogen concentrations.40 Moreover, inflammation-induced endothelial cell injury in different vascular beds may contribute to a hypercoagulable state and the risk of thromboembolic complications.41,42
To provide mechanistic insights into the reported hypercoagulable state of patients with severe COVID-19, we compared changes in the contact-phase system activation and fibrinolysis between patients with COVID-19, individuals with ARDS-influenza, and donors (healthy subjects). Whereas some critical parameters, such as fibrinogen, tPA, and TAFI, were significantly increased, FXII levels were reduced in severe COVID-19, and the process of fibrin formation and the resulting fibrin clot structure and lysis were substantially different between the patient cohorts. Histological data provided evidence of widespread, compact fibrin deposition in the lungs of patients with COVID-19, as opposed to those with ARDS-influenza.
In particular, the levels of FXII were decreased in patients with severe COVID-19, as compared with those with ARDS-influenza and donors, and FXII-activation products, such as cleaved HK and PKa-like activity, were altered in patients with SARS-CoV-2 infection. This scenario very likely reflects FXII consumption related to its autoactivation on negatively charged surfaces and its reciprocal activation by PKa. Decreased FXII levels in COVID-19 plasma are also in accordance with moderately elevated activated partial thromboplastin time values reported in other studies.43,44 The exacerbated consumption of FXII in severe COVID-19 is further supported by our in vitro experiments, in which the supplementation of COVID-19 plasma with exogenous FXII resulted in its rapid activation, presumably because of the presence of FXII autoactivation cofactors or increased PKa activity. Indeed, common pathological events observed in COVID-19, such as increased tissue cell stress with virus-mediated necrosis, endothelial dysfunction, and excessive neutrophil activation, lead to the release/exposure of large amounts of negatively charged molecules including NETs. NETs not only bind FXII but also serve as potent endogenous inflammation-dependent inducers of FXII autoactivation, eventually propagating thrombosis.45 Enhanced vascular NETosis along with impaired NET clearance have been described in patients with COVID-19.38,46 In line with these findings, several studies have found an increase in NET components in COVID-19 plasma, including cell-free DNA, myeloperoxidase-DNA complexes, neutrophil elastase-DNA complexes, and citrullinated histone H3.47,48 In addition, active FXII has been described to colocalized with NETs in the lungs of patients with COVID-19, and NET+ pulmonary vessels have been reported to be frequently clogged.38,49 Together with these findings, our results speak for NET-induced, accelerated, and constant activation of FXII in COVID-19 and thus for its role in immunothrombotic processes in this pathological condition. In fact, FXII autoactivation cofactors were found to be relevant for the initiation and progression of sepsis and DIC.50,51
Interestingly enough, low plasma levels of FXII in severe COVID-19 did not result in the prolonged lag phase in fibrin formation, triggered by kaolin, as opposed to that in ARDS-influenza. High levels of FVIII:C in the former group of patients may provide an explanation for this effect. However, further studies assessing the levels of other coagulation factors such as factor IX and XI and their inhibitors, as well as thrombin-antithrombin complexes, are needed to thoroughly characterize high procoagulant activity of COVID-19 plasma. In particular, Bouck et al recently reported high thrombin generation potential of COVID-19 as opposed to sepsis plasma.52 These results, together with previously described high levels of fibrinogen, mild thrombocytopenia, and slightly altered plasma concentrations of coagulation factors and physiological anticoagulants argue for a specific form of intravascular coagulation in severe COVID-19 that is distinguishable from the classical DIC in sepsis.53 The prominent increase in vascular complications points to strong involvement of endothelial cells in hemostatic abnormalities seen in COVID-19. Injured endothelial cells may provide a scaffold for thrombus generation and elevated levels of von Willebrand factor multimers (recently described in COVID-19 plasma54 ) may facilitate platelet-vessel wall interactions, ultimately leading to the formation of platelet-rich thrombotic deposits in microvasculature. Such platelet-rich thrombotic aggregates have been observed in alveolar capillaries of critically ill patients with COVID-19.16,17 Altogether, the hemostatic alterations in COVID-19 subjects reflect widespread occlusive thrombotic microangiopathy with destruction of alveoli that supports persistence of microthrombi.
Increased levels of fibrinogen and elevated thrombin generation potential contribute to the formation of stable clots composed of a dense network of thin fibrin strands.36,55 Accordingly, clots generated in COVID-19 plasma exhibit higher packing density and are more resistant to lysis, compared with clots formed in ARDS-influenza plasma. Further experiments with COVID-19 plasma revealed that next to fibrinogen, FXIIa may regulate clot compactness. Although increased fibrinogen levels can independently promote thrombus formation and stability,55 the role of FXII in this process seems to be more complex and dependent on the presence of the cofactors that enable FXII autoactivation. FXIIa may then regulate fibrin network density in a thrombin-dependent and -independent manner. Whereas the ability of FXIIa to convert FXI to FXIa and thereby to promote thrombin generation is well documented,56,57 elucidation of the mechanism of a direct effect of FXIIa on fibrin clot architecture requires further research. FXIIa binds with high affinity to fibrinogen/fibrin,36 whether this interaction facilitates fibrin fiber cross-linking or incorporation of other components into a clot is speculative at the moment. Although the high turbidity of fibrin clots observed in COVID-19 may speak for the intercalation of NET components into the fibrin network. Cell-free DNA and histones (both NET components) have been found to promote more opaque and fibrinolysis resistant clots.58,59 In addition, cell-free DNA has been reported to bind to fibrinogen, fibrin, FXII, FXIIa, HK, FIXa, FXIa, fibronectin, and von Willebrand factor.58,60-62 Thus, its intercalation into fibrin network may not only accelerate FXII autoactivation but also serve as a platform that brings plasma proteins and fibrin fibers together resulting in the formation of turbid and fibrinolysis-resistant clots.
The persistent vessel occlusion in critically ill patients with COVID-19 seems to be reinforced by markedly increased plasma levels of TAFI and moderately increased amounts of PAI-1. Elevated levels of tPA try to counterbalance this prothrombotic environment, but are not sufficient to compensate for increased procoagulant activity in patients with COVID-19. These findings are supported by recently published results showing that the enhanced thrombin generation potential is not adequately offset by increased plasmin generation potential in COVID-19.52 High levels of fibrinogen and accelerated rate of FXIIa formation seem to potentiate this effect.
Our study has several limitations. First, the number of patients with COVID‐19 or ARDS-influenza was small, and samples were collected at a single time point. Second, most of the patients with COVID‐19 or ARDS-influenza were receiving anticoagulant therapy. Five of the patients with ARDS-influenza and 7 of those with COVID-19 did not receive anticoagulation. However, we did not observe any differences in the parameters measured in the patients with or without anticoagulation. Third, although demographics and baseline laboratory values in ARDS-influenza and COVID-19 are comparable, there are some significant differences (eg, in CRP or procalcitonin levels) that may have had an impact on our results. Yet, exclusion of the ARDS-influenza cases with the highest CRP or procalcitonin values did not change our results. Fourth, blood samples used in this study were not supplemented with contact-phase pathway inhibitors, therefore, changes in the activation status of the contact-phase proteins during blood sampling cannot be excluded. However, all samples were collected in the same manner. Finally, the measurements of plasma function in ARDS-influenza and COVID-19 were performed in vitro; hence, they may not fully reflect the processes occurring in vivo. Thus, further mechanistic studies ex vivo and in vivo are needed to fully elucidate the procoagulant and fibrinolytic changes in ARDS-influenza and COVID-19.
In conclusion, pathological events described in COVID-19 create milieu favoring activation of FXII. In combination with high levels of fibrinogen, FXIIa may contribute to compact, lysis-resistant clot formation in a thrombin-dependent and -independent manner. This prothrombogenic microenvironment is further promoted by dysregulated fibrinolysis. Our study advances understanding of the common and divergent aspects related to clot formation and lysis during ARDS-influenza and COVID-19.
Acknowledgments
The authors thank E. Bieniek for excellent technical assistance and A. Seipp (Institute of Anatomy and Cell Biology, Justus Liebig University, Giessen, Germany) for obtaining the scanning electron microscopy images.
This study was funded by the German Research Foundation (DFG: SFB/TR84 Project A2) (M.W. and W.M.K.), the Else Kröner-Fresenius-Foundation (2014_A179) (M.W. and P.M.), the Oskar Helene Heim Foundation (P.M.), the German Center for Lung Research (82 DZL 005A1) (M.W.), and the University Medical Center Giessen and Marburg (UKGM) (M.W. and P.M.).
Authorship
Contribution: M. Wygrecka designed the study, performed experiments, analyzed data, and wrote the manuscript; A.B., L.M., and O.P. performed experiments and analyzed data; B.S., S.D., T.W., J.J.S., M.C.B., S.H., F.K., L.E.S., and M. Witzenrath recruited patients, analyzed clinical data, and reviewed the manuscript; A.-S.S. and F.S. analyzed clinical data and wrote the manuscript; M.Z. and G.G. collected autopsy tissue samples and reviewed the manuscript; N.W., R.T.S., G.B., L.S., and P.M. analyzed data and contributed to the writing of the manuscript; and W.M.K., G.K., and K.T.P designed the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
M. Wygrecka, B.S., T.W., N.W., R.T.S., G.B., P.M., M.C.B., S.H., F. K., L.E.S., M. Witzenrath, W.M.K., and K.T.P. are members of the German Center for Lung Research.
Correspondence: Malgorzata Wygrecka, Center for Infection and Genomics of the Lung, Universities of Giessen and Marburg Lung Center, Aulweg 132, 35392 Giessen, Germany; e-mail: malgorzata.wygrecka@innere.med.uni-giessen.de.

