Key Points
The VpreB component of the SLC is expressed in standard- and high-risk B-ALL.
The VpreB may serve as a novel immunotherapeutic target.

Abstract
Immunotherapies directed against B-cell surface markers have been a common developmental strategy to treat B-cell malignancies. The immunoglobulin heavy chain surrogate light chain (SLC), comprising the VpreB1 (CD179a) and Lamda5 (CD179b) subunits, is expressed on pro- and pre-B cells, where it governs pre–B-cell receptor (BCR)-mediated autonomous survival signaling. We hypothesized that the pre-BCR might merit the development of targeted immunotherapies to decouple “autonomous” signaling in B-lineage acute lymphoblastic leukemia (B-ALL). We used the Children’s Oncology Group (COG) minimal residual disease (MRD) flow panel to assess pre-BCR expression in 36 primary patient samples accrued to COG standard- and high-risk B-ALL studies through AALL03B1. We also assessed CD179a expression in 16 cases with day 29 end-induction samples, preselected to have ≥1% MRD. All analyses were performed on a 6-color Becton-Dickinson flow cytometer in a Clinical Laboratory Improvement Amendment/College of American Pathologist–certified laboratory. Among 36 cases tested, 32 cases were at the pre-B and 4 cases were at the pro-B stages of developmental arrest. One or both monoclonal antibodies (mAbs) showed that CD179a was present in ≥20% of the B-lymphoblast population. All cases expressed CD179a in the end-induction B-lymphoblast population. The CD179a component of the SLC is commonly expressed in B-ALL, regardless of genotype, stage of developmental arrest, or National Cancer Institute risk status.
Introduction
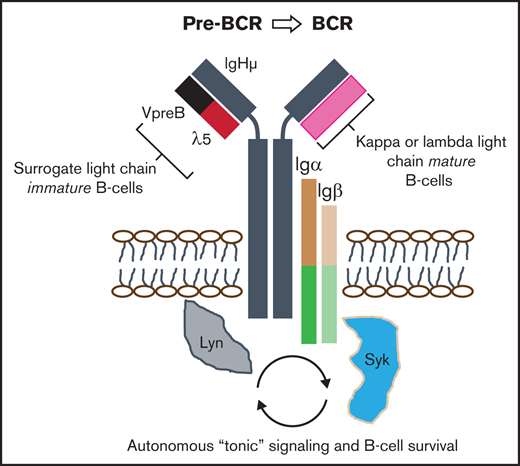
The productively assembled pre–B-cell receptor (pre-BCR) autonomously signals to govern immature B-cell selection and expansion into immunoglobulin-producing cells.1 The pre-BCR is composed of 5 units (see visual abstract): a membrane-bound V-, D-, J-recombined immunoglobulin heavy chain, an invariably constant surrogate light chain (SLC), comprising VpreB (CD179a) and λ5 (CD179b),1 and transmembrane immunoglobulin α (Igα) and Igβ accessory chains that coassemble to provide intracellular signaling through SRC and SYK family kinases.2,3 Differentiation into mature B cells can only occur when immature B-precursors have undergone recombination of genes encoding κ or λ light chains, which dynamically replace the SLC in maturing B cells to create a functional BCR.3 Without pre-BCR mediated “tonic” autonomous signaling, immature B cells undergo programmed cell death, but this critical selection step may be subverted by oncogenic transformation.1,4
Despite numerous genomic aberrations, nearly all B-lineage acute lymphoblastic leukemia (B-ALL) cases share a relatively restricted repertoire of B-cell surface markers, including CD19 and CD22, and with variable expression of CD34 or CD20.5-7 Despite the use of risk-adjusted therapies, relapse is a common problem for infants, adolescents, and adults.5,8 Novel immunotherapies have the potential to uncover unexpected escape pathways by which leukemic cells evade cell death.9 Although relatively little is known about the expression of the pre-BCR in B-ALL, others have concluded that the pre-BCR is functionally active in a small but important subset of ∼16% cases, designated “pre-BCR+ ALL.”7,10,11 For antibody-mediated therapy, surface expression, not signaling, mediates cell killing, as demonstrated by the efficacy of rituximab against many CD20-expressing neoplasms, including B-ALL.6
We recently described the characteristics of a novel high-affinity, high-avidity anti–pre-BCR antibody and evaluated whether blockade of homotypic pre-BCR self-associations might differentially sensitize primary patient samples to chemotherapy.2 We found that incubation of patient blasts with anti-VpreB monoclonal antibodies (mAbs) enhanced apoptosis by decoupling cell survival pathways. Because B-ALLs might resist cytotoxic therapies by means of autonomous survival signaling, we investigated whether CD179a, as an immunotherapeutic target, might be more commonly expressed on B-lymphoblasts than previously reported.10
Methods
To assess CD179a surface expression in B-ALL, we used National Cancer Institute (NCI) risk status and end-induction minimal residual disease (MRD) levels of ≥1% to select 36 diagnostic cases from Children’s Oncology Group (COG) Biology Study AALL03B1 (#NCT00482352) (supplemental Figure 1). To determine whether CD179a was expressed following a month-long course of induction therapy (supplemental Table 1), we obtained 16 paired, day 29 samples for further testing, 7 samples from standard-risk AALL0331, and 9 samples from high-risk AALL0232. All subjects and/or their legally authorized representatives provided written, informed consent in accordance with the Declaration of Helsinki. The study protocol was approved by the COG Cell Bank (AALL18B2-Q), Cancer Therapy Evaluation Program, and Children’s Minnesota IRB.
Samples were stained with 2 different antibody combinations (CD20–fluorescein isothiocyanate [FITC]/CD10-phycoerythrin [PE]/CD38-PerCPCy5.5/CD58-APC/CD19-PECy7/CD45-APCH7 and CD9/CD13+33/CD34/CD10/CD19/CD45), including a third tube with SYTO-16 to identify all nucleated cells (COG MRD Panel)12 that also included a PE-conjugated CD179a mAb (Biolegend, San Diego, CA). In cases where a paired, day 29 sample was available with sufficient viable cells for further sorting, a fourth tube was tested, which included a recombinant FITC-conjugated mAb against CD179a2 (produced by GenScript, Piscataway, NJ, with FITC-conjugation per the manufacturer’s instructions; Abcam, Cambridge, MA). Unlike the FITC-labeled conjugate, the PE-labeled conjugate does not undergo internalization (Biolegend, personal communication).2 CD3-PerCP, CD10-PE, CD13+/33-APC, and CD19-PECy7 were used as protocol controls (supplemental Table 1); positive and negative controls for the FITC-conjugated CD179a mAb were tested against Nalm6 cells (supplemental Figure 2). All analyses were performed on a Becton-Dickinson FACSCantoII 6-color cell analyzer in a Clinical Laboratory Improvement Amendment/College of American Pathologist–certified laboratory. Cases with ≥20% CD179a surface expression were determined to be positive for χ2 analyses; all comparisons were performed using GraphPad Prism 8.6 software (San Diego, CA).
Results and discussion
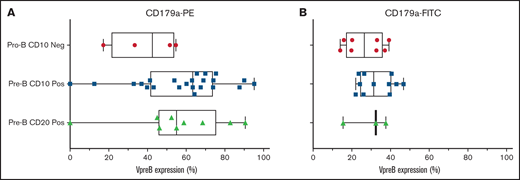
We evaluated 36 cases in total: 32 cases were arrested at the CD10+ pre-B stage and 4 cases were arrested at the CD10− pro-B stage (supplemental Table 2). We found no differences in VpreB expression among 3 developmental stages of B-ALL arrest (Figure 1; P = NS, 1-way analysis of variance). In diagnostic samples, VpreB-PE expression ranged from 0% to 95.2% (55.3% ± 3.9%), and for the FITC-conjugated mAb, expression ranged from 14% to 46.7% (mean, 29.6% ± 2.1%) (Figure 2A-B; unpaired Student t test, P < .001). One or both mAbs showed that CD179a was expressed in ≥20% of the B-lymphoblast population for all 36 diagnostic samples, including 3 cases for which RNA-sequencing data were available for comparative analyses (supplemental Table 3).13 Compared with earlier reports describing ∼16% of cases that expressed CD179a in B-ALL,10 we found that every case expressed CD179a in our series of 36 cases (2-sided Fisher’s exact test; P < .001). As described by Köhrer et al,10 we found CD179a expression in cases with E2A-PBX3 and KMT2A-R, but also with BCR-ABL1, consistent with Trageser et al,14 and in other genotypes that may present with ambiguous lineages.15 Because CD179a was ubiquitously expressed in our series of 36 cases, we hypothesize that it might be broadly targetable in B-ALL, including cases with elevated end-induction MRD.
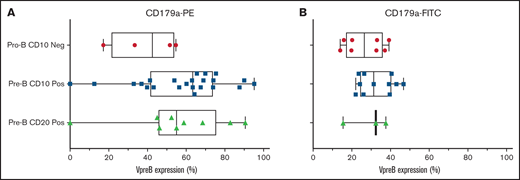
Using the VpreB-PE and the VpreB-FITC mAbs, 36 diagnostic cases were tested for VpreB surface expression in day 0 cryopreserved samples that were obtained from children and young adults with NCI standard- and high-risk B-ALL. Cases were subdivided into pro-B and pre-B-ALL based upon the absence or presence of coexpression with CD10 and CD20. There were no statistical differences in VpreB expression among these 3 subgroups, but all cases except 4 showed >20% expression using either the PE-conjugated (A) or the FITC-conjugated (B) CD179a mAbs. Lack of VpreB expression could not be correlated with the presence or absence of any recurring molecular aberrations, as shown in supplemental Table 2. Neg, negative; Pos, positive.
Using the VpreB-PE and the VpreB-FITC mAbs, 36 diagnostic cases were tested for VpreB surface expression in day 0 cryopreserved samples that were obtained from children and young adults with NCI standard- and high-risk B-ALL. Cases were subdivided into pro-B and pre-B-ALL based upon the absence or presence of coexpression with CD10 and CD20. There were no statistical differences in VpreB expression among these 3 subgroups, but all cases except 4 showed >20% expression using either the PE-conjugated (A) or the FITC-conjugated (B) CD179a mAbs. Lack of VpreB expression could not be correlated with the presence or absence of any recurring molecular aberrations, as shown in supplemental Table 2. Neg, negative; Pos, positive.
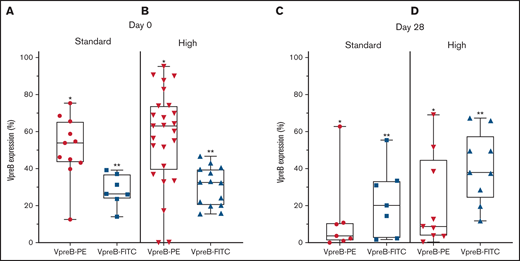
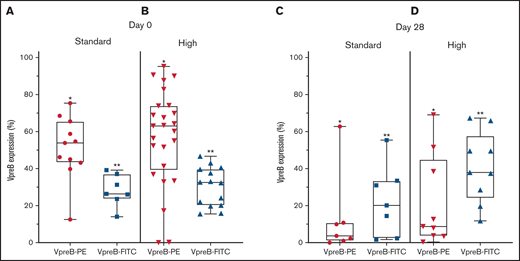
VpreB expression in standard- and high-risk B-ALL at diagnosis (day 0) and end induction (day 29). (A-B) VpreB-PE and VpreB-FITC in standard (A) and high risk (B) showed a spectrum of expression, but brighter expression for the PE conjugate (P < .001; unpaired Student t test) at the time of diagnosis. (C-D) VpreB-PE and VpreB-FITC in standard (C) and high risk (D) showed a spectrum of expression but trended to show brighter expression for the FITC conjugate (P < .001; unpaired Student t test) at end induction, suggesting that the FITC conjugate might detect recovering marrow populations that include B-lymphoblast populations with hematogones. Statistical comparisons were calculated between combined *PE and **FITC groups.
VpreB expression in standard- and high-risk B-ALL at diagnosis (day 0) and end induction (day 29). (A-B) VpreB-PE and VpreB-FITC in standard (A) and high risk (B) showed a spectrum of expression, but brighter expression for the PE conjugate (P < .001; unpaired Student t test) at the time of diagnosis. (C-D) VpreB-PE and VpreB-FITC in standard (C) and high risk (D) showed a spectrum of expression but trended to show brighter expression for the FITC conjugate (P < .001; unpaired Student t test) at end induction, suggesting that the FITC conjugate might detect recovering marrow populations that include B-lymphoblast populations with hematogones. Statistical comparisons were calculated between combined *PE and **FITC groups.
Because transient dimerization via the SLC is responsible for governing self-autonomous signaling in early B cells, we hypothesized that the abnormal MRD population might escape apoptosis following induction chemotherapy (supplemental Table 4; supplemental Figure 2). Using 2 CD179a-specific mAbs, embedded in the COG MRD panel, we found that both mAbs identified pre-BCR expression in an abnormal B-cell population (Figure 2C-D). For the PE-conjugated mAb, day 29 expression ranged from 0.4% to 69.9% (mean, 18.4% ± 5.9%), and for the FITC-conjugated mAb, expression ranged from 2.0% to 68.1% (mean, 33.4% ± 5.2%; P = .07). PE and FITC photon spectral intensities differ, the former fluorochrome having brighter emission with laser excitation at 488 nm.16 We speculate that variations in detectable pre-BCR expression between the 2 anti-CD179a mAbs may be attributable to differences between mAb specificity and sensitivity. The CD179a-FITC mAb showed greater detectable expression in the end-induction samples, suggesting that the mAb may have a differential sensitivity/affinity to B-lymphoblasts that co-mingle with an expanding pool of hematogones, which may also be present in a recovering marrow.17-19
B-ALLs relapsing after cell-based therapies demonstrate antigen remodeling, downregulation, lineage switches, and T-cell exhaustion.20 Relapsed or progressive disease in B-ALL may arise from a pervasive genetic/epigenetic reprogramming of B-lymphoblasts in what has been termed “senescence-associated stemness.”9 Because these changes are not reversed with the cessation of induction chemotherapy, relapse-initiating B-lymphoblasts exit senescence-associated stemness and establish therapy-resistant cell populations (identifiable as MRD), which then undergo clonal expansion, leading to relapse and death. These phenomena are especially common in KMT2A-Rs, BCR/ABL1, and other high-risk molecular lesions.15 By targeting the autonomous signaling controlled by the pre-BCR, it might be possible to bypass these cell-based mechanisms of relapse.
Because CD19 and CD22 are commonly expressed in B-cell neoplasms, these surface receptors have been logical targets for cell-based therapies (reviewed by Bonifant and Tasian).5,21 Trials utilizing rituximab have been successful, but have also been difficult to implement into clinical practice, because CD20 is uncommonly expressed in B-ALL.6 An important limitation for cell-based therapies against CD19, CD20, and CD22 immunotherapies in B-cell neoplasms is their elimination of all normal B cells; this leads to pan B-cell ablation with resultant immune dysregulation. In murine lambda5 and VpreB1/2 knockouts, mature B cells and immunoglobulins reach near-normal levels, suggesting mature B-cell sparing,22,23 and in humans, SLC expression is absent beyond B cells that express IgM.24,25 We therefore propose to assess in future clinical trials whether immunotherapies against the pre-BCR might spare a patient’s mature B-cell repertoire.
Acknowledgments
We acknowledge the support and assistance of David Teachey and Karen Rabin, cochairs of the COG Cell Bank, support and approval from the Cancer Therapy Evaluation Program section of the National Institutes of Health, and the Cancer and Blood Disorders section at Children’s Minnesota. A special word of thanks to the patients and families who supported this research through their consent to participate in COG ALL clinical trials AALL03B1, AALL0331, and AALL0232.
This work was supported by National Institutes of Health, National Clinical Trials Network (NCTN) Operations Center grant U10CA180886; NCTN Statistics and Data Center grant U10CA180899; COG Biospecimen Bank grant U24CA196173; NCI Outstanding Investigator Award NCI R35 CA197695 (C.G.M.); NCI Cancer Center Support grant CA021765 to St. Jude Children’s Research Hospital; American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital; and St. Baldrick’s Foundation.
Authorship
Contribution: S.S.W. and B.S.W. designed the research, analyzed the data, and wrote the paper; C.Q. and C.G.M. provided correlative RNA-sequencing experimental data from a larger study of 633 B-ALL samples; A.M. and N.S. performed the research, which was reviewed and analyzed by G.G.; and N.A.H., A.J.C., and B.L.W. contributed vital expertise to the data analysis and interpretation of COG clinical trial demographic data and its correlation with the experimental data.
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict-of-interest disclosure: The FITC-conjugated anti-VpreB antibody used in this study is protected by US patents 10,858,488 and, for therapeutic allowance, 10,988,533 (S.S.W. and B.S.W.). The remaining authors declare no competing financial interests.
Correspondence: Stuart S. Winter, Cancer and Blood Disorders, Children’s Minnesota, 2525 Chicago Ave South, Minneapolis, MN 55404; e-mail: stuart.winter@childrensmn.org.